Trombosi coronarica
Enciclopedia del Novecento (1984)
Trombosi coronarica
di Cesare Bartorelli e Maurizio Guazzi
SOMMARIO: 1. Introduzione. □ 2. Caratteristiche anatomiche e istologiche. □ 3. Complicanze d'ordine anatomico. □ 4. Meccanismi patogenetici. □ 5. Lo spasmo coronarico: a) lo spasmo coronarico come fenomeno biologico con significato clinico; b) meccanismi dello spasmo coronarico; c) spasmo coronarico e infarto miocardico. □ 6. Manifestazioni cliniche della trombosi coronarica acuta: a) segni fisici; b) rilievi enzimologici; c) altri rilievi umorali ed ematologici. □ 7. Diagnostica strumentale: a) elettrocardiogramma; b) ecocardiogramma; c) tecniche radioisotopiche. □ 8. Trattamento della trombosi coronarica acuta: a) misure terapeutiche d'ordine generale; b) interventi farmacologici rivolti alla limitazione della lesione ischemica. □ 9. Disturbi del ritmo e della conduzione: a) bradiaritmie; b) tachiaritmie. □ 10. Scompenso congestizio e shock cardiogeno nella trombosi coronarica acuta: a) interventi d'ordine farmacologico; b) interventi d'ordine meccanico; c) interventi d'ordine chirurgico. □ Bibliografia.
1. Introduzione.
Dal 1912, quando J. B. Herrick pubblicò l'articolo che ha per titolo Clinical features of sudden obstruction of the coronary arteries, la causa dell'infarto miocardico fu individuata in un'occlusione improvvisa di un vaso coronarico, di solito conseguenza di trombosi, e i termini ‛trombosi coronarica' e ‛infarto miocardico acuto' divennero sinonimi. Nonostante da oltre quarant'anni si sappia che questo concetto non risponde sempre a verità, dal momento che la trombosi coronarica può avvenire senza provocare infarto (v. Blumgart e altri, 1941) e che l'infarto può insorgere, come si dirà più avanti, per cause diverse dall'occlusione trombotica (v. Friedberg e Horn, 1939), la sinonimia ha persistito nel tempo ed è correntemente in uso anche ai nostri giorni. Per questa ragione essa sarà mantenuta anche nella stesura di questo capitolo.
Infarto miocardico significa necrosi delle cellule del muscolo cardiaco. Quasi tutti gli infarti del cuore traggono origine dall'aterosclerosi delle arterie coronarie. Aterosclerosi è un termine generale che identifica una o, più probabilmente, diverse forme di patologia vascolare. Essa sembra avere un substrato eziologico complesso e variabile, comprensivo di fattori genetici, metabolici e trombogenici e di stimoli stressanti, a loro volta interrelati con caratteristiche del comportamento, personalità, stati emotivi, preferenze dietetiche, dinamica circolatoria e livelli di pressione arteriosa sistemica. Indipendentemente dall'eziologia e patogenesi del processo aterosclerotico, il risultato finale è quello della formazione di placche che conducono al restringimento del lume coronarico. Come in altri letti vascolari, la presenza di aterosclerosi di per sé non riveste necessariamente un significato clinico o funzionale. Ciò che ha importanza è l'entità dell'ostruzione causata dalla lesione aterosclerotica e l'alterata dinamica circolatoria che da essa risulta, in relazione alla quantità di flusso ematico richiesto dal miocardio che da quel flusso trae la propria vitalità. Considerazioni energetiche fondamentali indicano che in assenza di flusso ematico la cellula miocardica diviene metabolicamente insufficiente nel giro di un singolo battito. Quando l'ischemia sia prolungata, si attua un danno irreversibile che, appunto, prende il nome di infarto cardiaco.
La sede e le dimensioni della zona infartuata dipendono da una serie di fattori, quali: localizzazione ed entità del restringimento del lume vasale, dimensione del letto vascolare perfuso dal vaso ostruito, grado di sviluppo dei vasi collaterali, necessità metaboliche del miocardio interessato dal processo ischemico.
2. Caratteristiche anatomiche e istologiche.
L'ostruzione della coronaria discendente anteriore di solito provoca infarto della regione anteriore e apicale del ventricolo sinistro, con possibile interessamento anche di porzioni del setto interventricolare, della parete laterale e inferoapicale. L'ostruzione del ramo circonflesso della coronaria sinistra può causare infarto della parete laterale o inferolaterale del ventricolo sinistro. Ostruzioni della coronaria destra in genere sono causa di infarto della parete inferoposteriore del ventricolo sinistro e delle porzioni inferiori del setto. Queste regole generali possono presentare eccezioni in relazione con la variabilità anatomica della distribuzione regionale dei vasi coronarici. Così, ad esempio, nel caso del ramo circonflesso dominante, la sua occlusione può provocare infarto, che si estende anche a livello parietale inferoposteriore del ventricolo sinistro. Quando, infine, una zona ventricolare sia quasi esclusivamente perfusa dal circolo collaterale, l'infarto può determinarsi a distanza dalla sede occlusa. Per esempio un'insufficienza graduale della coronaria destra può essere vicariata dallo sviluppo del circolo collaterale a partire dal ramo discendente anteriore della coronaria sinistra; un'occlusione di quest'ultimo vaso può così coinvolgere nel processo infartuale anche la parete inferiore del ventricolo sinistro.
Pur essendo il ventricolo sinistro la sede di gran lunga più frequente della lesione infartuale, è possibile un interessamento contemporaneo (25% dei casi: v. Isnar e Roberts, 1978) o isolato (2-3% dei casi: v. Roberts, 1972) del ventricolo destro. L'infarto ventricolare destro, sia esso combinato o isolato, è generalmente associato con lesioni ostruttive della coronaria destra. Tuttavia, esiste una chiara discrepanza tra frequenza di infarto con sede ventricolare destra e frequenza di interessamento aterosclerotico della coronaria omolaterale. Questa discrepanza può trovare una ragionevole giustificazione nel fatto che la richiesta energetica del ventricolo destro è, per evidenti ragioni funzionali, assai inferiore a quella del ventricolo sinistro: l'infarto del ventricolo destro, infatti, è più frequente nei casi in cui, per un motivo qualsiasi (stenosi della valvola polmonare, ipertensione del piccolo circolo, ecc.), la richiesta energetica di tale ventricolo è più elevata del normale. Un'altra possibile ragione è anche il fatto che il ventricolo destro, per il modesto spessore della sua parete, può, in parte, trarre nutrimento dal sangue della cavità ventricolare.
L'infarto atriale è un evento non frequente, e tuttavia possibile, che accompagna circa il 7% degli infarti che interessano il ventricolo sinistro (v. Wartman e Hellerstein, 1948; v. Wade, 1957). L'infarto atriale è meno frequente a sinistra, forse perché il sangue arterializzato che passa attraverso l'atrio sinistro provvede in parte al suo fabbisogno metabolico.
Quando la necrosi miocardica interessa a tutto spessore la parete ventricolare, l'infarto viene definito ‛transmurale'; quando invece la necrosi coinvolge soltanto gli strati sottoendocardici, o solo quelli intramurali, o entrambi, senza estendersi agli strati epicardici, l'infarto viene definito ‛non transmurale'. Nelle prime ore la zona infartuata non presenta modificazioni macroscopiche, ma entro 5-6 ore essa diviene pallida, bluastra e lievemente tumefatta; entro 20-30 ore si ricopre di essudato fibrinoso, per assumere, infine, entro 48 ore, un aspetto grigiastro dovuto all'infiltrazione granulocitaria. Nello spazio di 8-10 giorni il segmento parietale infartuato riduce il proprio spessore per la graduale rimozione del tessuto necrotico e assume via via un aspetto grigio gelatinoso fino a trasformarsi in tessuto cicatriziale, di colore bianco e di consistenza aumentata, che va progressivamente retraendosi nel tempo. Il processo cicatriziale inizia alla periferia dell'infarto e si muove in senso centripeto.
Le anomalie istologiche in genere manifestano le seguenti tappe evolutive: a distanza di 8 ore sono presenti edema dell'interstizio, deposito di materiale lipidico nelle fibrocellule e infiltrazione di granulociti (prevalentemente neutrofili) e di globuli rossi; dopo 24 ore si osserva la perdita della striatura delle fibrocellule e i nuclei divengono picnotici o, addirittura, scompaiono; durante i primi tre giorni il tessuto interstiziale diviene edematoso e ricco di globuli rossi; infine, a partire dal quarto giorno inizia la rimozione delle fibre necrotiche e si ha invasione di linfociti, macrofagi e fibroblasti che danno inizio al processo riparativo di granulazione, completo in genere verso la sesta settimana.
Le osservazioni ultrastrutturali raccolte in infarti sperimentali nell'animale suggeriscono che la prima anomalia, già rilevabile entro 20 minuti dall'occlusione coronarica, consiste in riduzione di dimensione e numero dei granuli di glicogeno e in rigonfiamento e distorsione dei tubuli del reticolo sarcoplasmatico e dei mitocondri. Lo schema più plausibile della sequenza degli eventi che hanno luogo nella cellula miocardica in conseguenza dell'ischemia è quello proposto da Ganote e altri (v., 1976): si realizza in definitiva un danno, compreso il rigonfiamento dei mitocondri, che può essere irreversibile. Se viene reinstaurato precocemente un flusso sufficiente, la cellula ritorna alla normalità e conserva una funzione normale; se l'ischemia persiste, la cellula risulta lesa in modo irreversibile, la permeabilità selettiva della membrana scompare e si ha fuoriuscita degli enzimi intracellulari. Se a questo punto viene ripristinato un normale apporto di ossigeno, si ha un rapido accumulo di calcio a livello del tessuto cardiaco, la cellula perde la funzione normale, presenta bande di contrazione a livello citoplasmatico e i mitocondri si arricchiscono di granuli di fosfato di calcio. Risulta evidente che l'integrità della membrana cellulare è un punto d'importanza critica durante lo stadio precoce dell'ischemia: una volta che essa sia compromessa, si accumulano ioni calcio entro la cellula e questa si avvia verso un danno irreversibile.
3. Complicanze d'ordine anatomico.
Sono rappresentate dalla rottura di muscolo papillare, del setto interventricolare e della parte non settale del ventricolo sinistro (la cosiddetta parete libera), dalla formazione di aneurisma parietale e dall'insorgenza di pericardite.
La rottura parziale o completa di muscolo papillare può interessare, con frequenza decrescente, il muscolo posteromediale di sinistra, quello anteromediale dello stesso lato e i muscoli papillari del ventricolo destro. La sezione completa di un papillare del ventricolo sinistro è un evento che mette in serio e imminente pericolo la sopravvivenza del paziente, essendo causa di incontinenza massiva della valvola mitrale, da cui deriva un altrettanto massivo ingorgo del letto polmonare. Edema polmonare intrattabile o shock cardiogeno sono in genere la causa di morte (v. Wei e altri, 1979). È, tuttavia, assai più frequente l'eventualità di una rottura parziale che, a sua volta, è causa di grave, ma non sempre fatale, insufficienza mitralica (v. fig. 1).
La perforazione del setto interventricolare è generalmente singola e può avere dimensioni variabili da pochi millimetri ad alcuni centimetri. E sempre secondaria a infarto transmurale localizzato o esteso alla parete settale. La conseguenza funzionale è quella del passaggio di sangue dal ventricolo sinistro a quello destro, da cui risulta un sovraccarico di volume del ventricolo sinistro. Quest'ultimo, infatti, si trova a dover accogliere in fase diastolica quell'eccesso di sangue di ritorno dal circolo polmonare che esso stesso ha rivolto verso questa nuova direzione. L'importanza funzionale, e quindi prognostica, del difetto è strettamente relata con le sue dimensioni anatomiche.
Fra le cause di morte, nella fase acuta dell'infarto miocardico, la rottura della parete libera del ventricolo sinistro ha un ruolo di responsabilità in circa il 10% dei casi (v. Bjorek e altri, 1972). Di solito questo evento porta all'inondazione ematica del pericardio e al tamponamento acuto del cuore. Esso è tipico degli infarti transmurali; ha una frequenza maggiore nel sesso femminile e una concentrazione temporale verso il terzo-quinto giorno dall'evento infartuale; è favorito dalla concomitanza di ipertensione arteriosa; ha come sede più frequente il territorio di distribuzione del ramo discendente anteriore della coronaria sinistra; in genere riconosce come meccanismo di formazione un ematoma dissecante che perfora una zona necrotica di miocardio; è relativamente raro in casi d'ipertrofia ventricolare sinistra. Quando l'ematoma dissecante rapidamente si avvia verso l'organizzazione e coinvolge in questo processo il pericardio, la lacerazione parietale può risultare tamponata e può così essere prevenuto lo sviluppo di emopericardio. Questa zona, col passare del tempo, può dar luogo a una formazione diverticolare più o meno grande, chiamata pseudo-aneurisma, che rimane in comunicazione con la cavità ventricolare attraverso uno stretto colletto. Tale condizione anatomica si differenzia dall'aneurisma vero in quanto la sua parete non contiene affatto fibrocellule miocardiche; dal punto di vista funzionale, tuttavia, un'importante somiglianza accomuna le due formazioni: entrambe accolgono una certa quota del volume sistolico, sottraendolo al flusso anterogrado verso l'aorta.
L'aneurisma ventricolare è costituito da una sacca o estroflessione della parete del ventricolo, che ha perduto la capacità contrattile perché formata da tessuto fibroso frammisto a un'insufficiente quantità di cellule vitali del miocardio. È una complicanza che caratterizza il 12-15% dei soggetti che sopravvivono all'evento infartuale (v. Abrams e altri, 1963). La formazione dell'aneurisma probabilmente avviene per stiramento della zona infartuata non contrattile, a cui segue assottigliamento della parete e invasione della medesima da parte di tessuto fibroso. Sotto il profilo funzionale questo segmento parietale, anziché accorciarsi e seguire un moto consensuale e centripeto nel corso della sistole, si espande in senso centrifugo alterando in modo sostanziale la dinamica e la potenza della contrazione ventricolare. Col trascorrere del tempo sulla parete aneurismatica possono avvenire depositi calcifici, talvolta così rilevanti da essere ben visibili radiologicamente e da delineare il profilo della sacca aneurismatica (v. fig. 2). Le sedi più frequenti sono l'apice e la parete anteriore del ventricolo sinistro. La rottura tardiva di un aneurisma ventricolare è un evento relativamente raro (v. Abrams e altri, 1963; v. Vlodaver e altri, 1975).
Quando l'infarto è transmurale, difficilmente il pericardio rimane indenne. Nel 50% dei casi esiste la possibilità di insorgenza, entro i primi 5 giorni, di pericardite regionale e nel 15% dei casi di pericardite diffusa, fibrinosa o siero-fibrinosa. La raccolta di siero nel sacco pericardico può essere anche cospicua, ma mai di entità tale da disturbare in modo importante la dinamica del cuore. Anche la sindrome di Dressier, o sindrome postinfartuale, si manifesta con pericardite regionale, ma con insorgenza più tardiva (verso la seconda-sesta settimana), ed è probabilmente il risultato di una reazione autoimmunitaria.
4. Meccanismi patogenetici.
Il tessuto miocardico diviene ischemico ogni volta che, per eccesso di richiesta o per difetto di apporto, la necessità energetica del cuore, propria di quel particolare momento, non viene adeguatamente soddisfatta. Come già è stato detto in precedenza, se la durata dell'ischemia supera un certo limite temporale, il danno cellulare diviene permanente e irreversibile: in altri termini, si attua quello che noi chiamiamo infarto. In teoria, quindi, l'infarto cardiaco potrebbe avvenire anche in presenza di un albero coronarico anatomicamente e funzionalmente normale, in seguito a una richiesta energetica miocardica prolungatamente superiore al limite delle capacità fisiologiche del flusso miocardico, quale può essere uno sforzo fisico spinto all'estremo. A parte questa eccezione, in altissima percentuale l'evento primario che conduce all'infarto è una patologia occlusiva del lume coronarico. Rimane un ristretto numero di casi in cui la necrosi miocardica non è la conseguenza di una lesione coronarica di tipo ostruttivo, ma di un possibile disturbo primario, qualitativo o quantitativo, del flusso coronarico (costrizione funzionale coronarica, ridistribuzione di flusso).
Le ostruzioni organiche (fisse) delle coronarie mostrano elevata variabilità; esse possono essere eccentriche o concentriche, brevi o lunghe, singole o seriate, possono ridurre il lume vasale in misura diversa e interessare un ramo arterioso dominante o non dominante. Le conseguenze emodinamiche di un'ostruzione coronarica sono determinate fondamentalmente dal grado di stenosi. Una riduzione dell'area di sezione pari o inferiore al 50% sembra avere scarso o nessun effetto sul flusso coronarico nella maggior parte delle circostanze applicabili ai fabbisogni circolatori dei Mammiferi. Le possibilità di incremento del flusso risultano significativamente diminuite quando la superficie di sezione del vaso è ridotta di circa il 70%. Importanti decrementi del flusso a riposo avvengono quando il lume vasale è ridotto in misura pari o superiore all'85% (v. Gould e Lipscomb, 1974); oltre questo limite, ulteriori e anche modesti decrementi del diametro intravasale causano rilevanti cali di flusso.
Un ruolo importante nell'accentuazione di un fenomeno ostruttivo, fino a giungere al limite estremo dell'occlusione coronarica completa, è stato assegnato a tre fattori: emorragia intraparietale, trombosi, alterata vasomotilità. Per quanto concerne il primo, è stato suggerito che i processi degenerativi intimali di origine aterosclerotica danneggino il tessuto di sostegno perivascolare e favoriscano la rottura di capillari con conseguente stravaso ematico intramurale. Questa emorragia può aumentare il volume della placca e occludere il lume arterioso o favorire la formazione trombotica. I meccanismi della trombosi sono ancora lontani dall'essere chiariti. Secondo una possibile interpretazione, l'ulcerazione di una placca aterosclerotica metterebbe a contatto diretto il sangue circolante con il collageno o altro materiale trombogeno parietale (v. Roberts, 1972). D'altra parte è pure vero che molti trombi possono sovrapporsi a placche aterosclerotiche con superficie integra. Per spiegare questa eventualità viene chiamato in causa l'intervento di forze attrattive piastriniche, tali da provocare aggregazione e conseguente formazione trombotica.
Anche se l'ipotesi della trombosi coronarica come causa diretta di infarto miocardico è accettabile su un piano logico ed è suffragata da una lunga serie di rilievi anatomici, essa è stata ed è oggetto di critiche e di discussioni che hanno dato origine a un'interpretazione esattamente opposta, vale a dire che la trombosi coronanca sia non la causa dell'infarto miocardico bensì la conseguenza, a sua volta favorita dal rallentamento o dall'arresto del flusso indotti dall'infarto. L'idea corrente (v. Erhardt, 1978) è che entrambi i meccanismi possono essere veri, sebbene in circostanze diverse e in pazienti diversi.
Per molti anni la vasomotilità e il possibile ruolo patogenetico del vasospasmo coronarico nell'ischemia e nell'infarto miocardico sono stati tenuti in ombra. L'interesse per questo aspetto della patologia coronarica e il relativo bagaglio di conoscenze sono andati crescendo, negli ultimi dieci anni, a un punto tale da far ritenere utile una trattazione a parte del cosiddetto spasmo coronarico.
Le variazioni dei fattori che influenzano la necessità di ossigeno del miocardio non sono, nel contesto dell'ischemia miocardica, meno importanti di quelle dei fattori, testé analizzati, che ostacolano il flusso coronarico. È stato dimostrato che gli aumenti della frequenza del battito cardiaco, della pressione di riempimento e svuotamento ventricolare e dello stato contrattile del cuore incrementano la necessità energetica del miocardio (v. Braunwald e altri, 19762). Ciascuno di questi fattori ha un effetto lineare positivo sulla richiesta di ossigeno del cuore. Sotto questo profilo sembrano avere importanza, pur se non perfettamente definita, anche il volume, la geometria, la rigidità e la complianza delle pareti ventricolari.
Infine, va menzionato il ruolo che hanno nella regolazione del flusso miocardico i fattori idraulici, estrinseci all'albero coronarico: la durata della diastole, la pressione diastolica aortica, il gradiente istantaneo esistente fra quest'ultima e la pressione diastolica ventricolare. Al proposito, basti ricordare come in presenza di vasi coronarici ostruiti, per aumenti anche lievi della pressione diastolica ventricolare o per riduzioni della pressione diastolica aortica, il gradiente effettivo di perfusione possa risultare limitato alla prima parte della diastole ed essere insufficiente a fornire agli strati muscolari subendocardici un flusso sanguigno adeguato.
Tutto questo rende ragione delle enormi differenze individuali fra i pazienti con malattia coronarica, in funzione della complessità della natura della malattia occlusiva e della varietà dei fattori che possono contribuire a renderla più o meno critica in termini dinamici.
5. Lo spasmo coronarico.
Per spasmo coronarico si intende un disordine della motilità coronarica tale da ridurre in modo importante il lume vasale e, pertanto, il flusso miocardico. Sotto il profilo clinico, lo spasmo coronarico può avere importanza nella misura in cui è in grado di ridurre la portata coronarica al di sotto di quel limite critico oltre il quale il miocardio diviene ischemico. Per il cardiologo quindi è importante conoscere non solo e non tanto se lo spasmo coronarico esista come fenomeno biologico, ma anche e soprattutto se esso abbia valore clinico in funzione dei suoi possibili rapporti con l'angina pectoris, l'infarto del cuore, la morte improvvisa. Nonostante i primi due di questi eventi abbiano come base comune l'ischemia miocardica, nella discussione dell'evidenza e del valore clinico dello spasmo coronarico è bene, per ragioni che risulteranno chiare da ciò che segue, che essi siano analizzati separatamente.
a) Lo spasmo coronarico come fenomeno biologico con significato clinico.
Il possibile ruolo di un'alterata vasomotilità coronarica nella patogenesi dell'ischemia miocardica e del suo sintomo soggettivo che va sotto il nome di angina pectoris, è stato oggetto di interesse e disputa per lunghissimo tempo (v. Latham, 1876; v. Osler, 1910; v. Gallavardin, 1973). Una serie di considerazioni suggerisce, quanto meno in modo indiretto, che lo spasmo coronarico sia estraneo all'angina di Heberden (v., 1772) che è la forma classica sollecitata dallo sforzo fisico. Osservazioni anatomopatologiche e studi coronarografici hanno dimostrato una frequentissima associazione di questa angina con lesioni organiche ostruenti almeno uno, ma di solito due o più grossi rami coronarici. In risposta a un aumentato lavoro del cuore, l'incremento dell'apporto di ossigeno dipende pressoché esclusivamente da un aumento della portata coronarica, essendo l'estrazione di ossigeno a livello miocardico massima anche a riposo. In presenza di un'ostruzione coronarica fissa compaiono ischemia miocardica e dolore anginoso quando la necessità di ossigeno del cuore supera la capacità di flusso del vaso ostruito (v. Gorlin, 1965); pressoché invariabilmente si osserva che l'insorgenza di angina è preceduta dall'aumento del battito cardiaco o della pressione arteriosa, o di entrambi, e che l'angina si dilegua dopo che frequenza e pressione sono di nuovo rientrate nei livelli basali. In un dato paziente i valori quantitativi di pressione e frequenza cardiaca che si associano con l'insorgenza di ischemia miocardica sono straordinariamente costanti, qualunque sia la causa (esercizio fisico, stress emotivo, ecc.) che sollecita la variazione circolatoria.
L'insieme di queste osservazioni ha portato alla teoria in base alla quale l'angina classica da sforzo trae origine dall'aumento della domanda miocardica di ossigeno in presenza di un flusso coronarico limitato. Una riduzione primaria e acuta del flusso, e quindi il vasospasmo coronarico, risultano non necessari ai fini interpretativi dell'ischemia miocardica da sforzo; pertanto non fu lasciato spazio a questa ipotesi che lo stesso Heberden (v., 1772) aveva prospettato nell'intento di interpretare la patogenesi dell'angina non da sforzo. La negazione del vasospasmo coronarico come fattore patogenetico dell'angina classica non ne escludeva, tuttavia, la possibile esistenza come fenomeno biologico. Va detto, anzi, che quest'ultima era suffragata dal rilievo di coronarospasmo in soggetti a lungo esposti, per ragioni professionali, all'azione della nitroglicerina e bruscamente sottratti alla medesima (v. Lange e altri, 1972).
I rapporti patogenetici fra disordini della vasomotilità coronarica e ischemia miocardica furono riallacciati circa vent'anni or sono, quando M. Prinzmetal e altri (v., 1959) descrissero una forma di angina che insorge a riposo, senza alcuna causa scatenante evidenziabile (apparentemente, quindi, non giustificabile sulla base di un aumento dello sforzo contrattile del cuore), per la quale essi prospettarono che il meccanismo responsabile fosse un temporaneo aumento del tono di un grosso ramo coronarico con lume ristretto da placca aterosclerotica. L'interpretazione di un critico calo di flusso come fenomeno primitivo - o, più precisamente, l'esclusione di un'aumentata richiesta energetica miocardica quale responsabile dell'ischemia - ricevette un solido sostegno dalla documentazione, ottenuta mediante monitoraggio elettrocardiografico ed emodinamico continuo e contemporaneo, che nel tipo di angina descritta da Prinzmetal nessuna variazione della frequenza cardiaca, della pressione arteriosa sistemica e polmonare, del volume minuto cardiaco, della pressione di riempimento dei due ventricoli e della durata della sistole precede e quindi determina l'ischemia (v. Guazzi e altri, 1971 e 1975). L'ischemia del miocardio è l'evento primario a cui si accompagnano, e di cui sono quindi consegnenza, importanti variazioni del circolo sistemico e polmonare (v. figg. 3 e 4) e della dinamica cardiaca (v. fig. 5), espressione di un grave difetto contrattile del cuore. Queste osservazioni incoraggiarono una lunga serie di indagini rivolte alla dimostrazione dell'angiospasmo coronarico come meccanismo scatenante dell'angina di Prinzmetal. Dal 1968 la letteratura medica si è arricchita di numerosissimi studi angiografici che documentano la riduzione del lume di un grosso vaso coronarico (fino all'occlusione completa) afferente al distretto del miocardio a cui elettrocardiograficamente corrisponde la sede dell'ischemia. La riduzione del lume vasale concomita col processo ischemico e si dilegua in pari tempo con il medesimo. Il restringimento del lume coronarico o la sua obliterazione completa sono stati osservati in vasi con lesioni evidenti e in vasi angiograficamente indenni. Sulla base di questi rilievi l'angiospasmo coronarico è stato insignito della dignità di fenomeno biologico con significato clinico e, in particolare, di meccanismo patogenetico dell'angina pectoris conosciuta come varietà di Prinzmetal (v. Ross e Gorlin, 1968; v. Dhurandhar e altri, 1972; v. King e altri, 1973; v. MacAlpin, 1973; v. MacAlpine altri, 1973; v. Oliva e altri, 1973; v. Linhart, 1974; v. Chahine e altri, 1975; v. Endo e altri, 1975; v. Maseri e altri, 1975; v. Gaasch e altri, 1976; v. Higgings e altri, 1976; v. Meller e altri, 1976; v. Wierner e altri, 1976; v. Selzer, 1977).
b) Meccanismi dello spasmo coronarico.
Trattandosi verosimilmente di un disordine vasomotorio, è comprensibile come sia stata tirata in causa per prima una disfunzione dell'innervazione o del sistema recettoriale dell'albero coronarico. Va ricordato che i vasi coronarici sono innervati da fibre sia simpatiche sia parasimpatiche (v. Hirsh e Borghard-Erdle, 1961) e sono forniti di recettori di tipo alfa (effetto vasocostrittore) e di tipo beta (effetto vasodilatatore), con netta prevalenza dei primi a livello dei vasi epicardici. Un'accentuazione del tono adrenergico esercita sulle arterie coronarie un azione prevalentemente vasocostrittrice; la stimolazione del ganglio stellato nel cane induce una costrizione coronarica che precede una vasodilatazione mediata da fattori metabolici (v. Zuberbuhler e Bohr, 1965). Esiste quindi un meccanismo fisiologico per la costrizione delle coronarie epicardiche a seguito di una stimolazione elettiva di tipo alfa. Tuttavia, in condizioni di normalità il flusso coronarico non dipende tanto da fattori di regolazione extracardiaci, quale il sistema neurovegetativo, quanto da fattori intrinseci al miocardio, che ne determinano il consumo energetico (v. tab. I). Ne consegue che il flusso coronarico - o, in altri termini, la vasomotilità - risente delle influenze neurogene non solo per effetto diretto, ma anche e soprattutto per effetto mediato (v. Gellai e altri, 1973), vale a dire in conseguenza delle variazioni di richiesta energetica che esse promuovono.

L'eccitazione dei recettori alfa è stata proposta come meccanismo del vasospasmo coronarico nell'angina di Prinzmetal da due gruppi di autori giapponesi, in base alla possibilità di sollecitare crisi anginose di questo tipo con l'uso di epinefrina (v. Yasue e altri, 1974 e 1976) o di metilcolina (v. Yasue e altri, 1974; v. Endo e altri, 1976) e, altresi, per l'osservazione che fenossibenzammina e atropina prevengono l'insorgenza di crisi anginose a opera dell'epinefrina e della metilcolina. L'azione alfa-stimolante di quest'ultimo farmaco, dotato di attività parasimpaticomimetica, si ritiene sia mediata dalla liberazione di noradrenalina a livello delle terminazioni simpatiche cardiache postgangliari (v. Blumenthal e altri, 1968; v. Levy, 1971). L'interpretazione neurovegetativa, tuttavia, è lungi dall'essere soddisfacente, perché nelle mani di altri ricercatori i metodi sopra menzionati di attivazione farmacologica dello spasmo coronarico si sono rivelati inefficaci (v. MacAlpin e altri, 1973) o, addirittura, i risultati clinici con l'uso di atropina (v. Meller e altri, 1976) sono stati opposti a quelli testé riportati. Va ricordato, altresì, che alla luce delle attuali conoscenze mal si giustifica un'eccitazione alfa-adrenergica selettiva al punto da interessare solo i vasi coronarici, senza alterare in alcun modo il circolo sistemico o la dinamica del cuore (l'ischemia, infatti, non è preceduta da alcun tipo di variazione emodinamica sistemica o cardiaca), e potente in misura tale da prevaricare i fattori vasodilatanti d'ordine metabolico attivati dal processo ischemico. Parrebbe più logico ammettere che si tratti di un'esagerazione non tanto dell'attività adrenergica, quanto della responsività dell'effettore; di quest'ultima, tuttavia, non esiste dimostrazione. Il fatto che il vasospasmo possa essere osservato sia in coronarie che presentino lesioni aterosclerotiche, sia in vasi angiograficamente indenni, è un ulteriore elemento di confusione.
Yasue e altri (v., 1978) hanno prospettato la possibilità che un' eccessiva disponibilità di ioni calcio nelle cellule muscolari lisce della parete delle arterie sia un anello importante della catena che porta verso lo spasmo delle medesime. La contrazione della muscolatura vasale è qualitativamente dipendente dalla presenza di ioni calcio, che sono necessari per l'attivazione dell'ATP-asi miofibrillare (v. Rüegg, 1971; v. Bohr, 1973; v. Fleckenstein e altri, 1976; v. tessuto muscolare). Gli ioni idrogeno sono potenti calcioantagonisti fisiologici, dal momento che competono con gli ioni calcio tanto a livello dei meccanismi di trasporto transmembranale, quanto a livello dell'ATP-asi miofibrillare (v. Mrwa e altri, 1974). Da ciò deriva che un eccesso di concentrazione di ioni calcio e un difetto di concentrazione di idrogenioni favoriscono la vasocostrizione, mentre un mutamento opposto della concentrazione dei medesimi promuove la vasodilatazione. Yasue e altri riuscirono a sollecitare coronarospasmo e attacchi di angina pectoris, in soggetti sofferenti di angina tipo Prinzmetal, abbassando la concentrazione ematica degli idrogenioni mediante iperventilazione e contemporanea infusione di soluzione alcalinizzante, e riuscirono inoltre a prevenirli con l'uso di un preparato calcioantagonista. Secondo gli stessi autori, l'angina a riposo, in particolare quella ricorrente nelle ore notturne, trova giustificazione nel calo della concentrazione idrogenionica che si ha durante il riposo fisico, specificamente durante quello notturno. Tale riduzione può significare sottrazione di un meccanismo competitivo verso lo ione calcio, suo aumento intracellulare, vasospasmo. Questa interpretazione, anche se lascia non chiariti diversi aspetti patogenetici dello spasmo coronarico e dell'angina a riposo, apre un orizzonte nuovo e forse più fecondo.
c) Spasmo coronarico e infarto miocardico.
Lo spasmo coronarico come possibile causa di infarto miocardico acuto è stato poco studiato. La ragione sta nel fatto che, tra le cause di infarto, l'interesse dei ricercatori è stato focalizzato pressoché esclusivamente sulla trombosi coronarica e sulla placca emorragica. Questi tre fattori, tuttavia, potrebbero essere interrelati: lo spasmo coronarico, in teoria, può favorire un rallentamento o un arresto di circolo e agevolare così la trombosi o la rottura di una placca aterosclerotica, seguita o meno da emorragia. Per contro, sempre su un piano ipotetico, la frammentazione di una placca o la trombosi potrebbero sollecitare l'angiospasmo. Alcuni dati di fatto rendono il ruolo dello spasmo coronarico meno remoto di quanto non si sarebbe portati a pensare. In 12 casi dei 32 originariamente descritti da Prinzmetal e altri (v., 1959) l'evoluzione fu verso l'infarto miocardico: in ciascuno di essi la sede infartuale fu la medesima in cui, durante le crisi di angina pectoris, avvenivano le modificazioni elettrocardiografiche transitorie, espressione di ischemia. Studi successivi hanno confermato questo rapporto. Assai significativo è il caso di un paziente (v. Johnson e Detwiler, 1977) in cui un infarto della parete anteriore del ventricolo sinistro seguì di tre mesi la documentazione di uno spasmo reversibile del ramo coronarico discendente anteriore che, in base ai rilievi angiografici, si presentava immune da lesioni aterosclerotiche. Esiste, altresì, una serie considerevole di casi di infarto documentato in modo inequivocabile, pur in assenza di anomalie coronariche evidenziabili con l'esame coronarografico.
Oliva e Breckinridge (v., 1977) eseguirono la coronarografia in 15 pazienti entro le 12 ore successive a un episodio infartuale acuto, prima e dopo l'iniezione intracoronarica di nitroglicerina. L'esistenza di spasmo coronarico sovrapposto a grave lesione aterosclerotica fu dimostrata con questo metodo in 6 soggetti (40% della casistica analizzata); in 5 di questi il tempo compreso fra l'inizio dei sintomi dell'infarto e l'esame angiografico non superava le 6 ore. Inoltre, Maseri e altri (v., 1975) hanno documentato che in pazienti il cui infarto è preceduto da episodi ripetuti di dolore anginoso a riposo, associati con sopralivellamento del tratto ST nell'elettrocardiogramma, l'infarto si associa con spasmo che in genere, ma non in tutti i casi, si sovrappone a una placca aterosclerotica.
Alla luce delle attuali conoscenze, l'interpretazione secondo cui lo spasmo coronarico osservato in questi casi sarebbe un evento secondario all'infarto ha probabilità di essere vera pari a quella secondo cui lo spasmo sarebbe l'evento primario. E possibile, quindi, che anche sotto questo aspetto i concetti fisiopatologici tradizionali relativi all'ischemia miocardica debbano essere riveduti.
6. Manifestazioni cliniche della trombosi coronarica acuta.
La sopravvivenza a un episodio infartuale acuto dipende dall'estensione della zona lesa e, in conseguenza o in parallelo a questa, dallo stato funzionale del cuore. La concomitanza di malattie addizionali, quali infarto miocardico pregresso, ipertensione, diabete mellito, e l'età avanzata del paziente costituiscono elementi aggravanti. I vari criteri prognostici che sono stati proposti, e che sono d'ordine clinico (congestione e rantoli polmonari), elettrocardiografico (estensione dell'onda Q a diverse derivazioni, persistente slivellamento del tratto ST e inversione dell'onda T), radiologico (ingrandimento dell'ombra cardiaca) ed enzimologico (livello ematico di creatinfosfochinasi e di transaminasi glutammico-ossalacetica), sono tutti quanti riconducibili ai due punti essenziali testé menzionati, vale a dire all'entità della lesione e al livello funzionale del cuore. Il fatto che l'infarto sia o non sia di tipo transmurale non sembra influenzare in modo significativo la prognosi. Anzi, va detto che la forma non transmurale può risultare un'entità instabile, in relazione sia alla sintomatologia anginosa, sia alla possibilità di estensione o ricorrenza dell'infarto.
Nonostante i progressi e l'ausilio diagnostico delle diverse metodiche biochimiche e strumentali, i sintomi o, ancor più genericamente, la storia riferita dal paziente rimangono il pilastro su cui fa perno la diagnosi. Non va dimenticato, tuttavia, che in un numero significativo di casi (intorno al 15%) l'infarto può decorrere totalmente asintomatico e la diagnosi può essere fatta a posteriori su rilievi elettrocardiografici o autoptici. Questi infarti, cosiddetti asintomatici, possono essere realmente tali (non si conoscono le ragioni dell'assenza del dolore) o possono falsamente risultare tali in funzione della breve durata della sintomatologia o della scarsa accuratezza posta dal paziente nel recepire, valutare e ricordare i propri sintomi fisici.
Nella maggioranza dei casi il dolore dell'infarto è persistente (di solito per periodi che vanno da mezz'ora a diverse ore), importante e spesso intollerabile. Esso può essere costrittivo, urente, oppressivo o trafittivo; la localizzazione è in genere a sede retrosternale con irradiazione simmetrica verso le parti alte del torace, fino al collo, o preferenzialmente a sinistra con estensione alla parte ulnare del braccio fino al polso. Altre sedi di interessamento dolorifico sono l'epigastrio (con possibile simulazione di patologia acuta addominale) e le regioni interscapolari con predilezione del lato sinistro. Gli eventi clinici più comuni che possono presentarsi con una sintomatologia dolorosa grossolanamente simile a quella descritta, ma diversa per molti dettagli, sono i seguenti: dissezione acuta dell'aorta, pericardite o pleurite acute, embolia polmonare, costocondrite o sternocondrite.
a) Segni fisici.
Il paziente con infarto acuto del cuore si presenta di solito ansioso, sofferente e in agitazione motoria, nel tentativo di trovare una posizione fisica idonea ad attutire il dolore. Quando la condizione funzionale cardiaca sia evoluta o stia per evolvere verso lo scompenso, il paziente presenta sudorazione fredda, posizione ortopnoica, difficoltà respiratoria, tosse ed espettorazione talvolta con striature ematiche.
La frequenza cardiaca può variare in rapporto con l'esistenza o meno di complicanze che interessino il ritmo e la conduzione cardiaca; in assenza di queste, la frequenza del battito è in genere elevata. La pressione arteriosa può essere moderatamente o, in casi isolati, marcatamente aumentata nella fase acuta dell'infarto; più spesso essa risulta ridotta anche a livelli estremi nel caso del cosiddetto shock cardiogeno. Per questa ragione, la pressione arteriosa dell'iperteso può scendere a livelli normali nel caso di infarto acuto, mantenersi tale per vari giorni e ritornare poi a valori elevati una volta iniziata o raggiunta la guarigione della lesione. La febbre, che si manifesta entro le prime 8 ore e rimane presente per la prima settimana, è un sintomo generale comune alla maggior parte degli infarti del cuore.
Se l'esame clinico viene condotto con oculatezza, è frequente il riscontro di uno smorzamento piùo meno marcato dei toni cardiaci, con possibile sdoppiamento del secondo tono. È di solito presente un quarto tono che trova giustificazione nella ridotta complianza e nell'aumento della pressione diastolica ventricolare. Quando è udibile anche un terzo tono, la complianza ventricolare è sicuramente ridotta in modo marcato e la funzione ventricolare compromessa. Questo segno, il cui rilievo è reso più agevole ascoltando il paziente in decubito laterale sinistro, ha un significato prognostico negativo per la condizione funzionale che riflette (v. Riley e altri, 1973). Un altro rilievo comune è quello di un soffio sistolico, transitorio o permanente, dovuto a dilatazione ventricolare sinistra o a disfunzione di muscolo papillare o a entrambi i meccanismi. Il soffio da rottura parziale o completa di muscolo papillare è sempre molto intenso, talvolta musicale e si associa costantemente a dispnea intensissima.
Rumori da sfregamento pericardico sono in genere un reperto comune dell'ascoltazione verso la seconda-terza giornata, ma possono avere durata fugace: proprio per questo, il loro riscontro risulterebbe più frequente se l'ascoltazione cardiaca dei soggetti con infarto transmurale fosse ripetuta sistematicamente nel corso dei giorni immediatamente successivi all'evento infartuale. Il riscontro tardivo di sfregamento pericardico (a distanza di alcune settimane dall'episodio acuto) rientra nel quadro della sindrome postinfartuale.
b) Rilievi enzimologici.
La determinazione dell'attività enzimatica nel sangue è divenuta importante per la diagnosi dell'infarto miocardico acuto. Nonostante l'indubbia utilità clinica dei test enzimatici, persistono tuttora difficoltà legate alla specificità, alla sensibilità e all'interpretazione. A partire dal 1954, quando J. S. LaDue e altri riportarono che l'enzima transaminasi glutammico-ossalacetica (SGOT) era aumentato nel siero di pazienti con infarto miocardico acuto, è stata indagata l'applicabilità clinica di molti test enzimatici per la diagnosi di infarto miocardico acuto. La pratica medica attualmente fa ricorso alla triade diagnostica di SGOT, latticodeidrogenasi (LDH) e creatinafosfochinasi (CK). L'uso clinico di questi test è basato su tre assunti: che la morte cellulare e non il semplice danno ischemico causi la liberazione di enzimi; che l'enzima circolante provenga dal cuore e non da altri organi; che esista una relazione diretta fra la massa del miocardio necrotico e l'attività enzimatica del sangue. Anche se nessuno di questi assunti è completamente provato, allo stato attuale la loro validità pare incontrovertibile.
Ciascuno di questi test enzimatici presenta vantaggi e inconvenienti che derivano, rispettivamente, da elevata sensibilità, intesa come frequenza con cui il test è positivo in presenza della malattia, e scarsa specificità, intesa come frequenza di una falsa positività (v. tab. II). La sensibilità (intesa come positività per 100 casi) della triade diagnostica, sempre che il sangue sia prelevato in modo appropriato entro le prime 36 ore, si avvicina al 100% se si fa riferimento a una casistica con conferma autoptica di infarto miocardico. Di conseguenza, se una sintomatologia dolorosa che richiama quella dell'infarto non si associa all'incremento di uno di questi tre enzimi, la diagnosi è in pratica esclusa. La specificità, invece, è ancora lungi dall'essere ideale, come risulta evidente dalla tab. II. Per questa ragione è bene che ciascun enzima sia discusso separatamente e siano indicate le possibili ragioni di interpretazione errata.
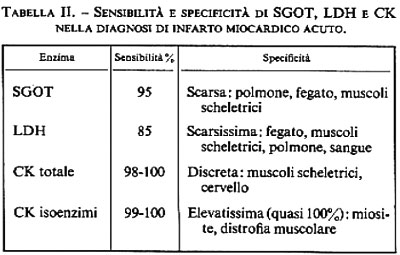
Per quanto riguarda la SGOT, va rammentato che con un suo aumento si associano scompenso di cuore, embolia polmonare, iniezioni endomuscolari, pericardite, malattie biliari e traumi chirurgici. La sua utilità clinica risiede, quindi, esclusivamente nella sua sensibilità. La LDH è meno sensibile e meno specifica della SGOT: la bassa specificità riconosce le medesime ragioni di quella della SGOT, ed è altresì in rapporto con l'elevata attività dell'enzima all'interno dei globuli rossi; ciò diventa quindi facile sorgente di errore quando vi sia emolisi e non perfetta preparazione dei campioni di sangue da analizzare. A seguito di infarto cardiaco la LDH rimane elevata per 6-8 giorni a causa del lungo tempo di clearance. Questo fatto torna utile in quei soggetti che hanno avuto infarto 48 ore prima della determinazione enzimatica, nei quali gli altri enzimi possono essere già rientrati nella normalità.
La CK è il più sensibile e il più specifico dei test enzimatici utilizzati nella pratica clinica. L'alta sensibilità deriva dalla considerevole attività specifica dell'enzima nelle cellule miocardiche; la soddisfacente specificità risiede nel fatto che l'enzima è limitato al cuore, ai muscoli scheletrici e all'encefalo, anche se è presente in piccole concentrazioni nel tubo gastroenterico, nel rene e nel polmone. L'attività della CK nel siero inizia ad aumentare 4-6 ore dopo il dolore cardiaco e raggiunge un picco entro 24-36 ore. Le iniezioni intramuscolari sono la causa più comune di falsa positività; seguono, in ordine di frequenza, distrofia muscolare, miopatie, cardioversione, traumi chirurgici, ipotensione, ipotermia, convulsioni, accidenti vascolari cerebrali. Va infine ricordato che l'attività fisica può far aumentare, anche se in modo non rilevante, l'attività della CK nel sangue e che, per questa ragione, sono indispensabili determinazioni seriate della CK ai fini di una diagnosi corretta di infarto miocardico.
Per aumentare sensibilità e specificità delle tecniche enzimatiche si è ricorsi allo studio del comportamento degli isoenzimi (v. enzimi). I vari organi si differenziano gli uni dagli altri non solo per i rispettivi tipi di enzimi, ma anche per le forme molecolari specifiche degli enzimi medesimi. Le diverse forme molecolari dello stesso enzima si chiamano isoenzimi. Per esempio, mentre il muscolo scheletrico utilizza un'ossidazione glicolitica e ha un isoenzima LDH che accentua la produzione di lattato, il muscolo cardiaco dipende da un metabolismo ossidativo piuttosto che da glicolisi e ha un isoenzima LDH che accentua la produzione di piruvato. L'utilizzazione del comportamento di questi isoenzimi ai fini diagnostici clinici ha per assunto che i profili isoenzimatici abbiano una specificità d'organo superiore a quella delle attività enzimatiche totali. È stato osservato, per esempio, che il cuore possiede un isoenzima CK relativamente specifico, che è l'MB-CK (v. Klein e altri, 1973). Questo isoenzima sembra essere un marker relativamente specifico del tessuto miocardico. A seguito di infarto acuto del cuore, l'MB-CK risulta aumentato nel siero 8-12 ore dopo l'inizio del dolore, se viene usato il metodo elettroforetico, e entro 3-8 ore se viene impiegato il metodo radioimmunologico. Il livello massimo raggiunto dipende dall'entità dell'infarto. L'attività MB-CK nel siero cessa più rapidamente dell'attività CK globale ed è di solito assente già 24 ore dopo il dolore. L'attività dell'isoenzima tornerà ad aumentare nel caso di estensione dell'infarto e, proprio per questo, essa viene considerata una spia sicura di questo evento. La sensibilità del test è intorno al 100%. La specificità è pure del 100% nei confronti del danno cellulare miocardico (lesione, flogosi, trauma, necrosi), mentre scende un po' al di sotto di questo livello per il puro infarto. Non si osserva incremento di MB-CK a seguito di iniezioni endomuscolari, trauma chirurgico, scompenso, ipotensione, ipotiroidismo, embolia polmonare. L'attività serica di questo enzima, quindi, sembra essere il test più sensibile e più specifico fra quelli attualmente disponibili per la diagnosi di infarto del cuore.
c) Altri rilievi umorali ed ematologici.
Si tratta di riscontri aspecifici, generalmente non utilizzati ai fini diagnostici, la cui conoscenza è, tuttavia, utile per evitare interpretazioni non corrette o errori diagnostici. L'infarto è frequentemente seguito da iperglicemia, anche in soggetti non diabetici. Azotemia e creatininemia possono aumentare in casi complicati da grave scompenso cardiaco con alterata emodinamica renale e ridotto filtrato glomerulare. Il quadro lipidico può risultare variato per la condizione di stress tipico dell'episodio infartuale. Entro poche ore dall'infarto può essere rilevata mioglobina in circolo. Il valore diagnostico della mioglobinemia è, tuttavia, assai inferiore a quello della CK per sensibilità e specificità. Un aumento del numero di globuli bianchi è riscontrato di frequente ed è interpretato come una risposta alla necrosi tessutale o all'aumentata secrezione di glicocorticoidi, o a entrambi. La leucocitosi è di tipo neutrofilo, si aggira intorno alle 15.000 unità per mm3, si manifesta entro poche ore, raggiunge un picco entro 2-3 giorni e scompare nell'arco di una settimana circa. La velocità di sedimentazione dei globuli rossi tende rapidamente ad aumentare, raggiunge un picco al quarto-quinto giorno e rientra nella normalità entro 3-4 settimane.
7. Diagnostica strumentale.
a) Elettrocardiogramma.
La legatura di un ramo principale di arteria coronaria nell'animale da esperimento è seguita da una serie di modificazioni elettriche che si susseguono secondo un certo ordine cronologico. Con la registrazione del potenziale d'azione transmembranale e di superficie è documentato (v. fig. 6) che una delle manifestazioni elettriche più precoci a livello della miocellula cardiaca dopo l'occlusione coronarica è un'accelerazione della ripolarizzazione, senza diminuzione del potenziale di membrana (v. Samson e Scher, 1964). A questo nell'elettrocardiogramma (ECG) di superficie corrisponde l'elevazione del tratto ST, che è comunemente indicata come onda di lesione. Col proseguire del danno ischemico si assiste a una progressiva riduzione di ampiezza del potenziale di membrana di riposo, che si traduce, nell'ECG di superficie, nella comparsa dell'onda Q e nella riduzione di voltaggio dell'onda R. Il comportamento elettrocardiografico nell'infarto umano è pressoché sovrapponibile e può, nella sua forma classica, essere così riassunto: soprelevazione convessa del tratto ST, inversione dell'onda T, onda Q anomala (con durata pari o superiore a 40 ms e voltaggio pari o superiore al 25% del voltaggio dell'onda R), associate con la depressione del tratto ST nelle derivazioni che esplorano la superficie non lesa del cuore (v. fig. 7).

L'elettrocardiogramma standard a 12 derivazioni è un mezzo di routine per la conferma o l'esclusione di infarto miocardico acuto. La correttezza diagnostica, sulla base dei soli rilievi elettrocardiografici, arriva all'80-90% dei casi, in particolare nella fase acuta dell'infarto. Questa percentuale può essere ulteriormente migliorata se i rilievi elettrocardiografici sono inseriti nel contesto clinico e affiancati ai dati enzimologici. L'accuratezza dell'elettrocardiogramma nella diagnosi dell'infarto può essere potenziata dall'esame vettocardiografico (VCG), che è di valido aiuto nei casi in cui i segni elettrocardiografici siano assenti, sfumati o comunque di dubbia interpretazione. A proposito di queste due metodiche, l'idea corrente è che l'uso combinato di ECG e VCG porti a risultati diagnostici migliori di quanto non avvenga con un loro uso separato; questo è particolarmente vero per certi tipi di infarto, quali sono quelli a sede strettamente posteriore. Per quanto si riferisce all'ECG standard a 12 derivazioni, può essere diagnosticato con certezza infarto miocardico solo quando siano presenti onde Q inequivocabilmente patologiche, accompagnate o no da altri segni elettrocardiografici già menzionati. Eccezioni a questa regola sono l'infarto ventricolare destro, quello atriale e quello a sede strettamente posteriore (v. tab. III). La localizzazione della sede infartuale viene determinata in base al numero e al tipo di derivazioni, fra le 12 dell'elettrocardiogramma standard, che presentino i segni caratteristici dell'infarto. Onde Q patologiche a cui non corrisponde infarto (cosiddetto comportamento pseudoinfartuale) possono essere osservabili in presenza di cardiomiopatia ipertrofica, blocco di branca sinistro, ipertrofia ventricolare sinistra e destra, sindromi di pre-eccitazione, accidenti vascolari cerebrali, anomalie della conformazione toracica, embolia polmonare. Va nuovamente ribadito, quindi, il concetto generale che la diagnosi elettrocardiografica di infarto deve essere inserita nel quadro clinico e suffragata dal medesimo.
b) Ecocardiogramma.
È nozione acquisita da molti anni che una delle manifestazioni più precoci dell'ischemia acuta o dell'infarto del cuore è un'alterata dinamica del segmento parietale il cui flusso ematico sia compromesso (v. Tennant e Wiggers, 1935; v. Prinzmetal e altri, 1949). L'ecocardiografla ha i requisiti per un utile impiego a fine diagnostico essendo una metodica incruenta, innocua e facilmente ripetibile, essa permette di valutare la dinamica del segmento parietale infartuato (v. fig. 8) e di seguirne l'evoluzione nel tempo. Tutto questo offre un ovvio vantaggio rispetto a metodiche tradizionali più invasive, quale la ventricolografia con mezzo di contrasto (v. radiologia medica).
L'ecocardiografia convenzionale (M-mode), che è stata applicata estensivamente allo studio della funzione ventricolare nella malattia coronarica, presenta, tuttavia, una certa limitatezza, dal momento che la possibilità di esplorazione è ristretta soltanto a piccoli segmenti del setto interventricolare e della parete posteriore del ventricolo sinistro. Pertanto, un certo numero di anomalie parietali, e persino aneurismi ventricolari, particolarmente quelli della parete anteriore, possono sfuggire all'indagine. Va inoltre ricordato che alcune variabili emodinamiche, quali volume sistolico, volume ventricolare e frazione di eiezione, possono essere calcolate in modo relativamente attendibile dalla semplice misura del diametro equatoriale del ventricolo sinistro, solo nel caso in cui morfologia, dimensioni e simmetria di contrazione parietale siano mantenute. Poiché l'ischemia e l'infarto del cuore alterano queste caratteristiche, l'utilità dell'ecocardiografia M-mode per la valutazione della prestazione ventricolare regionale e globale risulta ridotta in modo sostanziale in questo tipo di patologia.
Le limitazioni dell'ecocardiografia M-mode sono state per buona parte superate da una metodica più recente che va sotto il nome di ecocardiografia bidimensionale (EBD) in tempo reale, o scanning settoriale. Con questo metodo il cuore può essere esplorato sotto angoli di visuale diversi, tali da fornire una rappresentazione completa dell'organo. Rilievi sperimentali nell'animale hanno documentato che l'EBD può agevolmente localizzare e quantificare le anormalità di movimento parietale prodotte da occlusione coronarica acuta. Studi clinici condotti in fase acuta di infarto cardiaco hanno mostrato una chiara corrispondenza fra sede di anomalie dinamiche parietali e sede dell'infarto valutata con metodo elettrocardiografico. Va detto, anzi, che talvolta l'estensione della compromissione dinamica risulta superiore a quella predicibile sulla base dei soli rilievi elettrocardiografici. Una ragionevole spiegazione di questa discrepanza è che l'elettrocardiogramma standard offre un'esplorazione parziale delle diverse aree del miocardio e, quindi, non permette una definizione completa della superficie infartuata o dell'area ischemica peri-infartuale. E stata inoltre documentata l'esistenza di un parallelismo fra estensione ed entità del difetto dinamico parietale e gravità della condizione clinica del paziente (i soggetti con scompenso cardiaco o shock cardiogeno hanno invariabilmente anomalie di movimenti parietali più estese di quelle dei soggetti con infarto non complicato da questi eventi). Una buona correlazione è stata trovatà fra gravità della discinesia e livelli ematici di MB-CK, e fra evoluzione clinica ed evoluzione ecocardiografica della discinesia parietale.
L'insieme di questi dati conferma che l'indagine ecocardiografica, in particolare quella bidimensionale, è un utilissimo contributo nella valutazione diagnostica e prognostica dell'infarto miocardico nell'uomo.
c) Tecniche radioisotopiche.
Queste metodiche sono particolarmente interessanti per il cardiologo, perché sono praticamente non invasive, richiedono soltanto un'iniezione endovenosa della sostanza radiante e possono essere usate anche in pazienti con patologia cardiaca grave o acuta. Grazie alla disponibilità di gamma-camere mobili, le indagini radioisotopiche possono ora essere eseguite anche al letto del paziente e, quindi, impiegate senza rischio nelle unità per cure intensive coronariche in soggetti con infarto acuto del cuore. Le tecniche radioisotopiche in uso in cardiologia sono distinguibili in due gruppi quelle che permettono una valutazione della funzione ventricolare, in particolare della frazione di eiezione, e quelle che offrono una visualizzazione della perfusione miocardica regionale. Le une e le altre trovano utile impiego nel paziente colpito da infarto cardiaco.
La frazione di eiezione può essere misurata registrando, mediante gamma-camera connessa con calcolatore digitale, il primo passaggio attraverso il cuore del radionuclide iniettato in bolo per via endovenosa. Questo metodo, tuttavia, non permette una valutazione sufficientemente accurata della motilità regionale parietale. Per questo tipo di analisi è preferibile ricorrere al cosiddetto ECG-gated blood pool imaging, che consiste nella somministrazione endovenosa di un tracciante radioattivo con lunga semivita intravascolare, quali albumina e globuli rossi umani marcati con 99Tc. Dopo che la sostanza ha raggiunto una distribuzione ematica uniforme, l'immagine del cuore viene colta selettivamente in telesistole e in telediastole, ponendo il paziente in posizione obliquo-anteriore sinistra e obliquo-anteriore destra. Analogamente al metodo angiografico biplano, viene così tratteggiata l'immagine della cavità ventricolare sinistra, da cui è facile risalire ai volumi telediastolico e telesistolico, e da questi al volume sistolico e alla frazione di eiezione. Inoltre dal confronto delle due immagini non è difficile rilevare anomalie di movimento parietale, la cui definizione è resa ancor più agevole dalla registrazione delle immagini cavitarie ventricolari sinistre con cadenze intervallate da 40-60 ms, nel corso dell'intero ciclo cardiaco. Può essere così costruita una curva tempo-volume del ventricolo sinistro che permette un calcolo attendibile, oltre che della frazione di eiezione, della velocità massima e media di riempimento e di espulsione del ventricolo stesso.
La perfusione miocardica regionale è un'altra utile informazione offerta dalle metodiche radioisotopiche e può essere ottenuta mediante cold spot imaging o hot spot imaging. Per il primo metodo il radionuclide di scelta è il tallio 201, che si comporta biologicamente in modo simile al potassio (v. Bradley-More e altri, 1975) e, dopo essere entrato in circolo, si accumula a livello del muscolo cardiaco. La sua distribuzione iniziale in quest'organo sembra seguire molto da vicino la perfusione del medesimo (v. Strauss e altri, 1975). Pertanto, a livello delle aree parietali con flusso ridotto o assente, la captazione del tallio-201 risulta ridotta in modo evidente. Va detto inoltre che la captazione di questa sostanza a livello miocardico è in relazione con il trasporto transmembranale. Così, l'assenza o la riduzione di captazione può riflettere, oltre che un difetto di perfusione, un danno cellulare.
Il metodo hot spot imaging viene utilizzato per visualizzare l'infarto miocardico in fase acuta. Gli agenti impiegati per questo metodo hanno la prerogativa di accumularsi soltanto nel miocardio leso. Attualmente la sostanza utilizzata è il 99mTc pirofosfato: il meccanismo con cui questa sostanza va a depositarsi sul miocardio leso è ancora ignoto, né si sa se il deposito sia selettivo nel miocardio irreversibilmente leso o se venga interessato anche il tessuto danneggiato in modo reversibile. Le immagini scintigrafiche vengono in genere colte a distanza di 2 ore dall'iniezione. Nel soggetto normale la visualizzazione è soltanto a livello delle strutture ossee, nel soggetto con infarto acuto del cuore una discreta attività viene colta pure nella regione cardiaca e può essere localizzata in zone specifiche della parete ventricolare. Le migliori immagini si ottengono a distanza di 48-72 ore dall'episodio infartuale.
Le tecniche or ora descritte parrebbero avere un'ampia applicabilità nei pazienti con infarto acuto del cuore. Esse non vanno però considerate una routine, ma il loro impiego dovrebbe essere suggerito da precise necessità d'ordine conoscitivo e applicativo. Basterà citare, come esempi di utile indicazione, i seguenti casi storia clinica atipica, alterazioni elettrocardiografiche di non sicura interpretazione, infarto del ventricolo destro, anomalie elettrocardiografiche preesistenti, disturbi del ritmo e della conduzione che rendano dubbia la diagnosi elettrocardiografica, valutazione della funzione miocardica di base e in risposta a interventi farmacologici, estensione della zona infartuata e sua evoluzione nel tempo, reinfarto, shock cardiogeno, differenziazione fra insufficienza mitralica acuta e perforazione del setto interventricolare, valutazione prognostica in funzione dell'estensione della zona infartuata. A quest'ultimo proposito, va detto che immagini positive con 99mTc pirofosfato possono non essere completamente specifiche per l'infarto miocardico acuto, ma risultare positive, anche in tempi successivi, in casi in cui si sia sviluppato un aneurisma ventricolare, o in cui persista nel tempo un'evidente disfunzione parietale (v. Olson e altri, 1977). Questo aspetto, se da un lato rende meno specifico il test nei confronti della diagnosi di infarto acuto di cuore, gli attribuisce, dall'altro, il valore di indicatore altamente sensibile per la morbilità futura.
8. Trattamento della trombosi coronarica acuta.
Nella fase acuta dell'infarto la sopravvivenza è minacciata da due fattori: aritmie e scompenso cardiaco. Il 60% dei decessi avviene entro le prime ore dall'evento infartuale (quando in genere il paziente non ha ancora raggiunto un luogo idoneo per un adeguato soccorso) in conseguenza di aritmie maligne, in particolare di fibrillazione ventricolare. Nei tempi passati, anche dopo il ricovero in ospedale, aritmie e scompenso avevano pari responsabilità nel minare la sopravvivenza del soggetto. L'istituzione relativamente recente di ambienti (le cosiddette unità per cure intensive coronariche) idonei al monitoraggio continuo del ritmo cardiaco e alla messa in atto di efficaci misure di prevenzione e cura dei disturbi del ritmo e della conduzione ha fatto sì che, nei pazienti ricoverati in tali unità, il maggiore responsabile dei decessi sia oggi lo scompenso di cuore, lo shock cardiogeno in particolare. Solo una minoranza di decessi è ancora conseguenza di aritmie intrattabili, e queste di solito si associano con grave difetto dinamico del cuore.
La provata efficacia delle unità per cure intensive coronariche (UCIC) nella salvaguardia della vita del soggetto colpito da infarto, e la documentazione sicura che l'incidenza di aritmie minacciose è massima nelle prime ore dopo l'infarto e persiste, seppure con intensità decrescente, durante la prima settimana, hanno messo in chiara luce la necessità che il soggetto colpito sia affidato a cure specialistiche il più precocemente possibile e per una durata relativamente lunga. Da questo concetto ha tratto origine l'istituzione di unità mobili di cure coronariche per la fase che precede il ricovero nelle UCIC, e di unità di cure semintensive coronariche per la fase susseguente all'uscita dalle UCIC.
Le unità mobili sono costituite da autoambulanze dotate di un sistema di comunicazione via radio, di personale specializzato e di apparecchiature idonee per monitoraggio dell'elettrocardiogramma, defibrillazione e manovre di rianimazione, oltre che per la terapia medica del caso. Con questi mezzi il paziente può essere soccorso molto precocemente e ricevere i trattamenti necessari già durante la fase di trasporto all'ospedale. I rilievi statistici documentano che, dove esiste questo sistema di primo soccorso, la percentuale di decessi precoci è ridotta in modo rilevante.
Negli ultimi vent'anni, dopo l'avvento delle UCIC, la mortalità dei pazienti con infarto si è ridotta in misura oscillante fra il 30 e il 40%. Questi risultati significativi sono in massima parte riconducibili alla possibilità di porre rimedio ai disordini del ritmo e della conduzione; un certo merito, tuttavia, va riconosciuto anche ai sistemi di monitoraggio emodinamico attuabili in questi ambienti, che offrono una migliore guida per una terapia razionale dello scompenso.
L'esperienza clinica, i rilievi statistici e i dati sperimentali più recenti suggeriscono che una certa percentuale di morti improvvise (intorno al 10%) può avvenire per aritmie gravi in una fase relativamente tardiva, compresa fra la quinta e la decima giornata dopo l'infarto, in conseguenza di anomalie persistenti di vario tipo che interessano l'attività elettrica del miocardio leso e delle diramazioni più periferiche del tessuto di conduzione. Per questo, almeno in un certo numero di soggetti, si prospetta l'utilità di prolungare il monitoraggio del ritmo cardiaco anche dopo che il paziente è uscito dall'UCIC, fino alla settima giornata. Questo è quanto viene fatto nelle cosiddette unità per cure semintensive coronariche (v. rianimazione).
a) Misure terapeutiche d'ordine generale.
Il primo provvedimento terapeutico che dev'essere messo in atto è l'analgesia. Il farmaco da utilizzare è la morfina, somministrata per via endovenosa a dosi singole di 5 mg con intervalli di 10-15 ininuti, fino a quando il dolore dell'infarto non sia cessato o non si manifestino sintomi di tossicità. L'alleviamento del dolore è un punto critico nella terapia dell'infarto acuto, perchè rappresenta un mezzo per mitigare l'ansia e l'attività del sistema nervoso adrenergico.
L'abitudine clinica corrente di somministrare a ogni paziente colpito da infarto cardiaco ossigeno per le prime 24-48 ore non è probabilmente corretta, dal momento che non serve ad aumentare l'ossigenazione tessutale nel soggetto che sia già normossiemico, ma può favorire la vasocostrizione e il rialzo pressorio, effetti in genere non desiderati nella fase acuta dell'infarto. Più corretto è invece somministrare ossigeno solo in caso di necessità, tenendo come guida la pressione parziale d'ossigeno del sangue arterioso.
Se sia indicato o meno l'uso di anticoagulanti nella fase acuta dell'infarto è ancora argomento controverso. Nonostante gli oltre trenta studi clinici riportati dalla letteratura sull'argomento, da quando fu proposta nel 1948 la terapia anticoagulante, i pareri sono tuttora discordanti. L'origine prima di questo orientamento terapeutico sta forse proprio nella sinonimia, menzionata all'inizio, che lega i termini di infarto miocardico e trombosi coronarica. L'opinione ufficiale dell'American Heart Association, espressa da I. S. Wright (v. Wright e altri, 1948), era che l'uso di anticoagulanti poteva ridurre la frequenza di eventi fatali nell'infarto acuto del cuore. Per circa dieci anni, pertanto, fu considerato non corretto astenersi dall'uso di anticoagulanti nel paziente infartuato. Successivamente, con la comparsa nella letteratura medica di un numero sempre crescente di risultati che non dimostravano differenze sostanziali nella mortalità per infarto fra soggetti trattati e non trattati (v. Anticoagulants in acute..., 1973), l'uso degli anticoagulanti divenne l'eccezione piuttosto che la regola. Negli anni seguenti, tuttavia, il problema fu riproposto da due studi retrospettivi, pubblicati contemporaneamente nel 1975 (v. Modan e altri, 1975; v. Tonascia e altri, 1975), che suggerivano possibili effetti positivi.
L'atteggiamento corrente, basato su un'elementare ma valida razionalità, è che la terapia debba essere individualizzata. Anche all'occhio di chi non sia completamente addentro al problema balza evidente la differenza, per quanto concerne il pericolo di tromboembolia (trombi murali endocardici, trombosi venosa), fra un paziente con infarto non esteso, non complicato, per il quale la mobilizzazione dal letto può essere fatta in tempi precoci, e un paziente con bassa portata circolatoria (cardiomegalia, scompenso, shock), aritmie (fibrillazione atriale), stasi venosa o complicanze infettive che richiedano un prolungato soggiorno in letto. La pratica attuale è proprio quella di riservare alla seconda categoria di soggetti la cura anticoagulante. Questi ricevono in genere, a scopo profilattico, eparina e successivamente anticoagulanti orali. Una pratica che sembra sempre più diffusa è quella dell'impiego di antiaggreganti piastrinici, quali sulfinpirazone (v. Sulfinpyrazone in the prevention..., 1978), acido acetilsalicilico, dipiridamolo.
Il paziente con infarto cardiaco non complicato può iniziare a lasciare il letto dopo 36 ore dall'evento, passando in poltrona circa mezz'ora due volte al giorno. Al terzo o quarto giorno, se il decorso è tranquillo, egli può essere trasferito dall'unità per cure intensive all'unità per cure semintensive coronariche, per altri tre-quattro giorni. Sempre in assenza di complicanze, il paziente può iniziare a camminare nella stanza di degenza verso il sesto-settimo giorno. L'attività fisica può aumentare gradualmente nei giorni successivi. Un riposo a letto eccessivamente prolungato può, specialmente nell'anziano, favorire una serie di complicazioni, fra cui stipsi, ulcere da decubito, decalcificazione ossea, embolie polmonari, decondizionamento alla posizione eretta. La stipsi va accuratamente evitata, perché lo sforzo fisico di un'evacuazione difficoltosa può ridurre il ritorno venoso diminuendo esageratamente, in una condizione circolatoria precaria qual è quella dell'infarto acuto, la portata circolatoria sistemica e la perfusione coronarica, e favorendo l'insorgenza di aritmie. La correzione di anomalie del pH ematico, squilibri elettrolitici, tossicità da farmaci, ipovolemia, fa parte delle misure terapeutiche d'ordine generale.
Infine, il calore umano del personale, medico e non, e la sua disponibilità nell'assistere il paziente e nello smorzare l'impatto emotivo dell'evento fanno parte integrante della terapia dell'infarto acuto di cuore.
b) Interventi farmacologici rivolti alla limitazione della lesione ischemica.
È inequivocabilmente evidente che la prognosi di chi va incontro a infarto miocardico è relata in massima parte con l'estensione del tessuto cardiaco sano e potenzialmente vitale da cui dipende la funzione ventricolare residua. Pertanto, è stata prospettata la possibilità che le conseguenze immediate e tardive dell'infarto acuto siano influenzate in senso favorevole limitando l'estensione del danno ischemico nel momento della presunta occlusione coronarica. La base razionale su cui poggiano i tentativi di contenere il danno ischemico deriva dall'opinione che le aree del miocardio nel territorio del vaso occluso siano inizialmente esposte a pericolo, ma non irrevocabilmente compromesse. Per questa ragione la preservazione di queste aree mediante il ripristino di una perfusione adeguata o mediante la diminuzione della loro necessità di ossigeno è stata considerata un auspicabile traguardo terapeutico.
L'impiego dei farmaci bloccanti dei recettori beta-adrenergici cardiaci (v. sistema nervoso autonomo) è stato da più parti proposto come un mezzo utile a questo fine. Facendo riferimento alla tab. I, si comprende come questi agenti diminuiscano il consumo di ossigeno del miocardio (MVO2) riducendo la pressione arteriosa sistemica, la frequenza e la contrattilità del cuore. Essi, d'altro canto, tendono ad aumentare il consumo medesimo favorendo l'incremento delle dimensioni diastoliche del cuore e la durata della fase espulsiva ventricolare. Il risultato globale, tuttavia, è in genere una limitazione della richiesta di ossigeno da parte del cuore: su questa base poggia l'impiego dei beta-bloccanti nella cura dell'angina pectoris. D'altra parte, la dimostrazione sperimentale che i farmaci che tendono ad aumentare l'MVO2 incrementano il danno ischemico miocardico, e quelli che riducono l'MVO2 hanno un effetto protettivo, ha suggerito la possibilità che i beta-bloccanti influenzino favorevolmente il corso delle conseguenze ischemiche di un'occlusione coronarica acuta. In contrasto con l'abbondanza dei dati raccolti nell'animale da esperimento, che attestano l'efficacia del blocco beta-adrenergico nel proteggere il miocardio ischemico, il progresso degli studi clinici in questo settore è stato relativamente lento. Uno studio di H. S. Mueller e altri (v., 1974) ha documentato che il propranololo, somministrato per via endovenosa a dosi ripetute di 0,1 mg per chilogrammo di peso corporeo, migliora la condizione metabolica dei soggetti colpiti da infarto, riducendone il consumo di ossigeno e la produzione di lattato, senza provocare danni importanti d'ordine elettrico o dinamico. Questi risultati ebbero conferma in studi successivi di C. T. Peter e altri (v., 1978), con l'ulteriore osservazione che a distanza di 4 ore dall'episodio infartuale la liberazione globale di CK, la velocità di liberazione e il livello massimo dell'enzima erano, rispettivamente, inferiori del 30, 38 e 33% nei soggetti trattati rispetto a quelli non trattati. Tuttavia, nessuna differenza statisticamente significativa in questi indici era rilevabile fra i due gruppi se l'indagine veniva effettuata oltre 4 ore dopo l'episodio infartuale. Utilizzando come test il comportamento dell'elettrocardiogramma, H. K. Gold e altri (v., 1976) documentarono una significativa riduzione, dopo propranololo, dell'onda di lesione (sopralivellamento del tratto ST) nelle derivazioni precordiali di soggetti colpiti da infarto e trattati entro le prime 9 ore. A questi effetti elettrici si associava una consistente riduzione del dolore toracico prodotto dall'infarto. Queste osservazioni furono interpretate come espressione di un effetto benefico sul danno ischemico. A risultati simili hanno portato gli studi di Waagstein e Hjalmarson (v., 1976) che hanno utilizzato beta-bloccanti di tipo cardioselettivo.
I risultati di questi studi, seppure fortemente suggestivi, non possono e non debbono essere considerati conclusivi per le ragioni che seguono. Primo, esiste qualche perplessità circa il fatto che le variazioni degli enzimi e dell'ECG riflettano realmente ed esclusivamente le modificazioni di estensione e gravità del processo lesivo; secondo, manca una documentazione convincente che alla riduzione di estensione dell'infarto corrisponda un miglioramento funzionale del cuore; terzo, non esiste finora la prova che l'effetto apparentemente benefico dell'intervento farmacologico sia correlato con una riduzione della mortalità a breve o a lungo termine. La dimostrazione di questa correlazione rimane un punto cruciale prima che l'uso dei beta-bloccanti possa essere raccomandato come misura abituale per la riduzione dell'ischemia nel contesto dell'infarto acuto del miocardio. Non andrà però dimenticato che questi preparati potranno essere utilizzati impunemente solo in pazienti con infarto non complicato, senza manifestazioni di difetto dinamico del cuore.
Un altro gruppo di farmaci impiegati nell'intento di proteggere il miocardio ischemico è quello dei vasodilatatori. A questo proposito va ricordato che la distensione e la dimensione diastolica del ventricolo (preload) e il carico contro cui esso si contrae (afterload) influenzano in modo diretto la sua tensione parietale e quindi l'MVO2. I vasodilatatori possono influenzare il bilancio energetico del miocardio sostanzialmente in tre modi: riducendo il preload, riducendo l'afterload, modificando il flusso coronarico. Fatta eccezione per i cosiddetti calcioantagonisti (verapamil, nifedipina, ecc.) che, insieme a un'azione vasodilatante coronarica e sistemica, hanno un effetto farmacologico specifico d'ordine elettrico e metabolico a livello del miocardio, i vasodilatatori comunemente intesi non esercitano un'azione diretta di tipo inotropo o cronotropo sul cuore, e quindi non ne influenzano in modo diretto il bilancio energetico.
La riduzione del preload con agenti vasodilatatori, in particolare quelli con azione predominante sul distretto venoso, ha i presupposti per esercitare un effetto benefico sull'area ischemica sia riducendo la tensione parietale, sia aumentando la perfusione miocardica a segnito di incremento del gradiente diastolico transmurale. È probabile che quest'azione benefica sia più evidente in casi di pressione diastolica ventricolare esageratamente elevata, anche se non va trascurato il fatto che un'accentuazione del preload è talvolta un meccanismo indispensabile per mantenere la prestazione miocardica a livelli compatibili con la sopravvivenza. Parimenti, mentre è inconfutabile che una riduzione dell'afterload abbassa il consumo di ossigeno del miocardio e migliora la prestazione del ventricolo che tende allo scompenso, è altrettanto vero che la concomitante riduzione della pressione aortica può portare la perfusione coronarica al di sotto del livello ottimale. Questa considerazione si addice particolarmente all'infarto miocardico acuto, dove i meccanismi di autoregolazione perdono di efficacia e la perfusione coronarica può risultare relata in modo lineare con il livello di pressione arteriosa sistemica. Da questi rilievi, per quanto concerne la riduzione dell'estensione e della gravità del processo lesivo, parrebbe che i pazienti con infarto acuto che possono trarre i migliori benefici da un intervento con vasodilatatori siano quelli con livelli elevati non solo di pressione di riempimento ventricolare, ma anche di pressione arteriosa sistemica. Occorre ricordare, inoltre, che il mantenimento di una normale perfusione coronarica è cruciale nelle primissime fasi dell'infarto, mentre, già a distanza di 6-8 ore dall'episodio infartuale, la vitalità della zona lesa può essere poco o nient' affatto influenzata. È pertanto improbabile che interventi farmacologici con vasodilatatori o con altri mezzi esercitino influenze positive sul miocardio ischemico, se il rimedio viene attuato oltre un certo intervallo temporale.
Meritano ancora qualche considerazione teorica gli agenti calcioantagonisti, cioè quei farmaci che rallentano o bloccano la penetrazione transmembranale e la liberazione intracellulare dello ione calcio a livello della fibrocellula miocardica e vasale. Questa azione si traduce in un decremento dell'attività contrattile del cuore (interferenza negativa con l'accoppiamento eccitazione-contrazione) e in un effetto miorilassante sulle pareti vasali coronariche e sistemiche. Pertanto, l'effetto finale di tipo cardiovascolare osservabile in vivo è la risultante di una complessa interazione di variazioni simultanee di preload, afterload, contrattilità, frequenza cardiaca e flusso coronarico. A questo effetto finale, che generalmente si traduce in una diminuzione dell'MVO2, va affiancata la possibilità che il sovraccarico intracellulare di calcio abbia un ruolo di responsabilità nella patogenesi di un danno ischemico irreversibile (v. Jennings e Ganote, 1974; v. Kloner e altri, 1974) e che i calcioantagonisti esercitino un'interferenza positiva. È già stato ricordato, infatti, che la riperfusione di una zona miocardica irreversibilmente lesa dal processo ischemico è seguita dal deposito di fosfato di calcio a livello mitocondriale. D'altra parte, osservazioni sperimentali in vitro (v. Nayler e altri, 1976) e in vivo (v. Reimer e altri, 1977) documentano che agenti calcioantagonisti riducono l'estensione del danno ultrastrutturale del cuore reso ipossico e susseguentemente riperfuso.
L'interesse per questo aspetto farmacologico particolare dei calcioantagonisti, che li differenzia dai vasodilatatori comuni, è ovvio. Tuttavia, occorre ancora tempo prima che sia disponibile un'evidenza sperimentale solida al punto da garantirne un impiego clinico nell'infarto acuto non complicato.
Ritornando all'uso dei vasodilatatori classici (quali nitroglicerina e nitroprussiato di sodio) nella fase acuta dell'infarto nell'uomo, va fatta una chiara distinzione fra la provata utilità come rimedio allo scompenso e l'efficacia nel ridurre il danno ischemico. Per quanto concerne quest'ultimo aspetto, è stato dimostrato sperimentalmente che i vasodilatatori possono migliorare il metabolismo del miocardio reso acutamente ipossico; mancano, invece, dati convincenti riguardo agli aspetti istologici e istochimici.
Sotto il profilo clinico, non è sufficiente provare o aver provato che i vasodilatatori determinano modificazioni favorevoli nell'emodinamica cardiaca, nella liberazione degli enzimi cellulari, nella mobilità parietale, nelle immagini scintigrafiche, ma è indispensabile disporre di prove inconfutabili che il loro uso precoce migliora la morbilità e la mortalità dell'infarto. Questa prova allo stato attuale non esiste. Il ricorso indiscriminato a questi farmaci nell'infarto acuto al fine di proteggere il miocardio ischemico è, quindi, per ora criticabile; il loro uso potrà essere limitato ai casi che presentino elevata pressione di riempimento ventricolare sinistro associata con alti livelli di resistenza vascolare sistemica.
9. Disturbi del ritmo e della conduzione.
I disturbi del ritmo e della conduzione del cuore in corso di infarto miocardico possono essere i più disparati. In questa sede verranno trattati quelli di più comune riscontro e di maggiore rilevanza clinica.
a) Bradiaritmie.
La bradicardia sinusale è il disturbo del ritmo più comune nelle prime ore che seguono l'evento infartuale ed è preferenziale per gli infarti a sede inferiore. Se, da un lato, essa può avere un risvolto favorevole perché riduce il consumo di ossigeno del miocardio, dall'altro può essere causa di ipotensione e di ipoperfusione coronarica. Il rallentamento del battito è in genere la conseguenza di due ordini di fattori: 1) accentuazione dell'attività vagale mediata da recettori cardiaci a sede elettiva nella porzione posteroinferiore del ventricolo sinistro (v. Thoren, 1976); 2) disfunzione del nodo del seno o ischemia atriale. Il meccanismo vagale è in genere responsabile delle bradicardie che si manifestano entro le prime 4-6 ore, mentre la disfunzione del nodo del seno o dell'atrio è di solito alla base delle bradicardie più tardive. Le prime vengono risolte con l'uso di atropina (l'indicazione all'intervento farmacologico è un calo eccessivo della pressione arteriosa sistemica), le seconde di solito non richiedono trattamento. In quest'ultimo caso, tuttavia, se la condizione clinica lo rendesse necessario, il ritmo cardiaco verrà aumentato mediante stimolazione elettrica, piuttosto che con interferenza farmacologica con l'attività vagale.
L'asistolia ventricolare è causa di morte improvvisa. Generalmente è la conseguenza di fibrillazione ventricolare o di anomalie di conduzione atrioventricolare o intraventricolare. Il primo caso richiede defibrillazione elettrica, il secondo necessita di stimolazione elettrica immediata tramite l'applicazione di catetere transtoracico. Poiché la fibrillazione ventricolare è la causa di gran lunga più frequente di asistolia, è raccomandabile ricorrere, in prima istanza, alla defibrillazione elettrica in tutti quei casi in cui il meccanismo dell'asistolia ventricolare rimanga dubbio.
Altre cause di bradiaritmia sono i blocchi atrioventricolari e i blocchi intraventricolari. Il danno ischemico può produrre blocchi a livello del nodo atrioventricolare (blocchi A-V), della branca destra o sinistra del fascio di His, del fascicolo anteriore o posteriore in cui si divide la branca sinistra (emiblocco anteriore o posteriore sinistro). Nello stesso paziente può essere presente più di una di queste anomalie, in varia combinazione. I blocchi A-V vengono così definiti: a) di primo grado, quando tutti gli impulsi generati a monte dei ventricoli passano ai ventricoli medesimi impiegando un intervallo di tempo superiore al limite della normalità (200 ms); b) di secondo grado, quando solo una parte degli impulsi raggiunge i ventricoli; c) di terzo grado (o blocco completo), quando nessun impulso originato al di sopra dei ventricoli raggiunge i ventricoli medesimi e questi si contraggono per un impulso che ha origine al di sotto degli atri. Il blocco A-V di secondo grado viene distinto in tipo I e tipo II. Nel tipo I il blocco è a livello del nodo atrioventricolare (o nodo di Tawara) ed è caratterizzato dal fatto che il tempo di passaggio degli impulsi elettrici dagli atri ai ventricoli diviene sempre più lungo, fino a giungere all'interruzione del passaggio di un impulso, dopodiché il ciclo ricomincia e si ripete nel tempo con il medesimo comportamento. Nel tipo II il difetto di conduzione è quasi sempre a valle del nodo atrioventricolare ed è caratterizzato dal blocco improvviso della trasmissione di un impulso atriale ai ventricoli, senza che alcun allungamento della conduzione atrioventricolare preceda il blocco medesimo. Il blocco A-V di tipo II può avere cadenza variabile o seguire un regolare ritmo di comparsa.
Il blocco A-V di primo grado si manifesta nei pazienti con infarto acuto del cuore in circa il 10% dei casi. Di solito non richiede un trattamento specifico; tuttavia, se il blocco si associa a un marcato rallentamentò del battito e a ipotensione (espressione di esagerata attività vagale) può essere indicato l'uso di atropina. Neanche il blocco A-V di secondo grado tipo I richiede intervento terapeutico, sempre che non siano presenti bradicardia esagerata, scompenso congestizio, shock: in questi casi è indispensabile l'attivazione immediata di uno stimolatore (pace-maker) elettrico. Il tipo II è relativamente raro nell'infarto acuto ma, quando è presente, può essere il segno premonitore di un blocco di terzo grado. In questi casi è norma cautelativa tenere inserito un catetere venoso per la stimolazione elettrica, data la possibilità di evoluzione verso il blocco completo.
La stimolazione elettrica è assai raccomandabile in tutti i pazienti con infarto acuto complicato da blocco A-V di terzo grado. Essa rappresenta un mezzo protettivo contro possibili e deleteri disturbi emodinamici derivanti da bradicardia eccessiva o da asistolia ventricolare. La prognosi, comunque, nei casi con blocco A-V completo è seria, poiché si tratta in genere di una complicanza di infarti estesi della parete anteriore.
I blocchi intraventricolari (della branca destra o sinistra, o di uno dei fascicoli di divisione della branca sinistra, isolati o in combinazione) si manifestano in circa il 15% dei casi di infarto acuto. Essi influenzano in modo sensibilmente negativo la prognosi del paziente, non solo e non tanto per il difetto di conduzione in sé, quanto per l'entità del danno miocardico di cui questi blocchi sono conseguenza. È documentato che gli emiblocchi anteriori sinistri si associano con la prognosi meno grave. L'unico intervento terapeutico attuabile nei blocchi intraventricolari è l'applicazione di pace-maker, da attivare nel caso in cui il disturbo di conduzione evolva verso il blocco completo. Il pace-maker dovrà essere mantenuto cronicamente in quei soggetti in cui nella fase acuta dell'infarto il blocco intraventricolare sia stato complicato da un transitorio blocco completo. La ripetizione di quest'ultimo anche in fase tardiva è, purtroppo, una realtà frequente. Va ricordato, tuttavia, che anche la fibrillazione ventricolare è una causa non rara di morte improvvisa in questi soggetti.
b) Tachiaritmie.
La tachicardia sinusale è presente in circa il 30% dei casi di infarto acuto, ed è in genere la conseguenza di un'eccitazione dell'attività adrenergica che deriva dal dolore, dallo stato emotivo, o da più complessi meccanismi fisiopatologici (v. Malliani e altri, 1969). Un eccesso di catecolammine a livello del miocardio ischemico, se da un lato può risultare utile nel sostenerne l'attività dinamica, dall'altro può essere deleterio per l'incremento del consumo di ossigeno e per gli effetti aritmogenici che le catecolammine esercitano in queste particolari condizioni. L'associazione di tachicardia sinusale, circolo iperdinamico ed extrasistolia ventricolare è una condizione in cui i bloccanti dei recettori beta-adrenergici cardiaci trovano indicazione elettiva.
Fra le tachiaritmie con origine sopraventricolare la fibrillazione atriale occupa un posto importante, non solo per la frequenza relativamente elevata con cui si manifesta nell'infarto acuto del cuore (10-15%), ma anche per le conseguenze negative d'ordine emodinamico (viene, infatti, a mancare il desiderabile effetto della contrazione atriale nel mantenere una sufficiente pressione di riempimento ventricolare) e per la sua frequente associazione con lo scompenso cardiaco congestizio e con importanti lesioni parietali anteriori. Anche il flutter atriale, che è relativamente raro (1-2%), in genere si associa con un grave difetto dinamico del cuore e riveste un significato prognostico pari a quello della fibrillazione atriale. Entrambi questi tipi di aritmia sono più comuni durante le prime 24 ore e tendono di frequente a ripetersi anche dopo il ripristino più o meno prolungato del ritmo sinusale. Il loro trattamento dev'essere immediato, sia per le ragioni emodinamiche già menzionate, sia per gli effetti negativi che la tachiaritmia ha sul consumo energetico del cuore. Il primo scopo da raggiungere è il rallentamento del battito ventricolare e per questo la digitale è il farmaco di elezione, che offre inoltre il vantaggio di sostenere l'attività dinamica del ventricolo, in genere, come già detto, compromessa. Quando lo scompenso domina il quadro clinico, è raccomandabile fare ricorso alla cardioversione elettrica.
La cardioversione o la stimolazione atriale rapida sono solitamente i rimedi da adottare per la tachicardia parossistica sopraventricolare, dopo che si siano rivelate inefficaci le manovre di attivazione vagale (manovra di Valsalva, compressione dei seni carotidei, ecc.). La frequenza di questo tipo di aritmia nell'infarto cardiaco non supera l'1-2%.
Nel gruppo delle tachiaritmie di origine ventricolare sono comprese le extrasistoli, la tachicardia e la fibrillazione venticolare. Le extrasistoli ventricolari (altrimenti dette battiti prematuri ventricolari) si manifestano nella quasi totalità degli infarti del cuore. Il loro significato clinico varia a seconda della sede, della frequenza e delle modalità di insorgenza, ed è primariamente improntato alla possibilità di evoluzione verso la fibrillazione ventricolare. Sotto questo profilo, vengono considerate significative o minacciose quelle extrasistoli che si ripetano con frequenza superiore a 5 per minuto, specialmente se accoppiate o a salve, abbiano configurazione variabile e compaiano nella fase precoce della diastole (tali da interessare l'onda T della sistole precedente) durante il periodo di maggiore vulnerabilità elettrica del ventricolo (v. fig. 9). Il ruolo ‛premonitore' di questo tipo di aritmie nei confronti della fibrillazione ventricolare rimane, tuttavia, relativamente incerto, poiché si è osservato di frequente che la fibrillazione ventricolare può svilupparsi anche in assenza delle medesime e che, d'altra parte, molti casi di infarto non evolvono verso la fibrillazione ventricolare pur in presenza di esse. Nella fase tardiva dell'infarto, invece, il rischio di morte improvvisa sembra più strettamente relato con la presenza di extrasistoli ventricolari che presentino le caratteristiche sopra descritte.
Nonostante la complessità e la non completa definizione dei rapporti fra extrasistoli ventricolari e fibrillazione ventricolare, l'opinione corrente è che le extrasistoli minacciose debbano essere opportunamente trattate. Nella fase acuta dell'infarto il meccanismo dell'extrasistolia, più che a una aumentata automaticità, sembra essere riconducibile al meccanismo del rientro. Sotto questo profilo un ruolo importante è certamente quello dei fattori adrenergici. Per questa ragione, in particolare se concomitano tachicardia sinusale e circolo iperdinamico, il trattamento d'elezione è quello con beta-bloccanti, sempre che per essi non esista una specifica controindicazione clinica. Nel caso in cui non sia evidente una iperattività adrenergica, il farmaco da utilizzare è la lidocaina, da iniettarsi per via endovenosa in boli ripetuti, o da infondersi in continuo per la stessa via. Ovviamente, dovrà essere posta precisa attenzione nell'evitare di raggiungere dosi tossiche del farmaco, tali da provocare iperattività nervosa centrale e depressione della conduzione e dell'attività contrattile del cuore. La percentuale di successi della lidocaina contro l'extrasistolia ventricolare, da circa il 70% dei casi nella fase precocissima dell'infarto, passa al 90% nelle fasi più tardive, probabilmente in virtù dell'azione elettiva del farmaco sull'automaticità, che viene a essere il meccanismo predominante nella genesi dei battiti prematuri ventricolari. Nei casi in cui l'attività contrattile del cuore risulti significativamente depressa può essere impiegata, in alternativa alla lidocaina, la procainammide. Un terzo farmaco di provata efficacia è la fenilidantoina che, tuttavia, a differenza di lidocaina e procainammide, non offre sufficiente protezione contro la fibrillazione ventricolare nel caso in cui questa tenda a manifestarsi nonostante la soppressione dell'extrasistolia ventricolare.
La tachicardia ventricolare è correntemente definita come tre o più battiti ectopici ventricolari che si susseguono con frequenza superiore a 120 per minuto. Questa aritmia, la cui incidenza nell'infarto acuto e intorno al 20%, si associa in genere con lesioni transmurali estese e grave compromissione dinamica del cuore, e richiede un intervento terapeutico immediato che sarà diverso a seconda della frequenza ventricolare. Se questa supera i 150 battiti per minuto, è bene ricorrere all'applicazione di shock elettrico; se è di entità inferiore e ben tollerata dal punto di vista emodinamico, potrà essere fatto un tentativo iniziale d'ordine farmacologico con lidocaina o procainammide. Questi due farmaci, indipendentemente dal mezzo utilizzato in prima istanza per interrompere l'aritmia, dovranno essere somministrati per un tempo sufficientemente lungo a garantire che la medesima non si ripeta.
La fibrillazione ventricolare (v. fig. 10) interessa il 10% dei casi e in genere si manifesta in fase precocissima di infarto, entro le prime 4-8 ore. Manifestazioni assai più tardive (nel corso delle prime 4-5 settimane) sono particolarmente possibili in soggetti con infarto anterosettale complicato da blocco di branca destro o sinistro. La fibrillazione ventricolare nell'infarto si definisce primaria quando avviene in modo repentino, in soggetti con sintomi di scompenso scarsi o nulli, preceduta o no da battiti prematuri ventricolari premonitori. Si definisce secondaria quando è l'evento finale di una condizione di scompenso progressivo e irreversibile. Nonostante la gravità e la potenziale letalità della forma primaria, l'esperienza clinica ha insegnato che, se prontamente trattata, essa può non influenzare in senso negativo la prognosi. La forma secondaria, invece, proprio perché associata con grave compromissione anatomica e funzionale del cuore, ha una prognosi infausta in un'altissima percentuale di casi. L'unico rimedio possibile ed efficace è lo shock elettrico applicato con la massima urgenza. Tanto più lungo è il tempo che intercorre fra insorgenza di fibrillazione e ripristino di una valida e ordinata contrazione ventricolare, tanto minori risultano le possibilità che quest'ultimo abbia luogo, e tanto più elevate le probabilità di un danno ischemico cerebrale irreversibile. Il successo della defibrillazione è facilitato, e la possibilità di ripetizione è ridotta, dalla somministrazione contemporanea di bretilio tosilato, che possiede una sicura attività antifibrillatoria. La normalità dell'equilibrio acido-base e un'adeguata assistenza ventilatoria sono requisiti indispensabili affinché la defibrillazione abbia successo e affinché questo sia duraturo. Quando la manovra defibrillatoria abbia ripristinato una normale attività elettrica dei ventricoli, ma questa sia dissociata da un'efficace attività dinamica, esistono fondate ragioni per ritenere che la lesione infartuale sia molto estesa o sia complicata da perforazione settale o rottura della parete libera del ventricolo sinistro.
10. Scompenso congestizio e shock cardiogeno nella trombosi coronarica acuta.
Lo shock cardiogeno dovuto a carenza dinamica del ventricolo sinistro è la causa predominante di morte per infarto miocardico nei pazienti ospedalizzati. Il difetto fisiologico fondamentale alla base dello scompenso congestizio e dello shock nell'infarto cardiaco è una depressione dell'attività contrattile conseguente alla perdita di muscolo cardiaco funzionante. Una compromissione importante della dinamica del cuore ha come ovvia conseguenza l'abbassamento del livello di perfusione dei vari organi e sistemi. Il difetto funzionale cardiaco in genere si traduce in una depressione di tutti i parametri che riflettono lo stato contrattile, vale a dire pressione sistolica, volume minuto, volume sistolico, lavoro cardiaco e frazione di eiezione, e in un aumento della pressione di riempimento ventricolare. Sebbene ciascuna di queste variabili sia in genere alterata già in presenza di infarto non complicato, il grado di compromissione è più evidente in presenza di scompenso cardiaco (inteso nel senso classico), ed è massimo quando si raggiunge la condizione di shock. Quest'ultima condizione, quindi, non è altro che uno stadio molto avanzato dello scompenso congestizio, in cui l'elemento dominante non è tanto la congestione polmonare, quanto l'ipoperfusione tessutale. L'evoluzione verso lo shock è relativamente rara negli infarti non transmurali e in quelli transmurali a sede inferiore, diviene più frequente negli infarti transmurali a sede anteriore e raggiunge la massima incidenza negli infarti transmurali che interessano contemporaneamehte la parete anteriore e quella inferiore. In circa la metà dei casi i sintomi dello shock hanno un inizio precoce, entro poche ore dall'infarto. In questi casi l'indagine ecocardiografica e scintigrafica evidenzia una discinesia parietale anteriore e apicale che di solito interessa una porzione superiore al 50% della totalità del ventricolo sinistro. Tutto questo è generalmente la conseguenza di un'occlusione prossimale e completa del ramo discendente anteriore della coronaria sinistra. In una certa percentuale di infarti che evolvono verso lo shock, elementi precipitanti sono l'instaurarsi di insufficienza mitralica, di perforazione del setto interventricolare e di tamponamento cardiaco. Una manifestazione tardiva (vari giorni dopo l'episodio infartuale) è in genere tipica di soggetti con cardiomegalia, con infarto pregresso, o comunque con manifestazioni cliniche di scompenso già esistenti prima dell'episodio infartuale.
Un appropriato monitoraggio emodinamico in questi casi è un presupposto utile e forse indispensabile per un approccio terapeutico razionale e per una valutazione prognostica. Il fatto che le probabilità di regressione dello shock siano tanto più elevate quanto più precoce è l'intervento terapeutico conferma l'utilità di un monitoraggio emodinamico, grazie al quale possono essere colti i segui premonitori dello shock. I parametri circolatori più significativi al proposito sono: la pressione arteriosa sistemica (misurata con metodo intravasale), la pressione di riempimento dei due ventricoli, il volume minuto cardiaco, la resistenza vascolare sistemica e l'indice di lavoro del cuore
Lo scopo della terapia è quello di raggiungere un flusso e una pressione di perfusione tali da soddisfare le richieste sistemiche, migliorare la funzione cardiaca e prevenire l'estensione dell'infarto. Una prima misura terapeutica è costituita dall'espansione della massa circolante che, di per sé, può migliorare il volume minuto cardiaco e la pressione sistemica o, anche se inefficace sotto questo profilo, può portare a livelli ottimali il volume ematico e la pressione di riempimento del ventricolo sinistro prima che sia dato inizio a una terapia specifica di tipo farmacologico o di altro tipo. L'espansione della massa circolante può incrementare il volume minuto cardiaco aumentando il ritorno venoso al cuore e, per conseguenza, la distensione diastolica di quest'ultimo. Il miglioramento della funzione di pompa poggia, in questo caso, sulla relazione di Starling che, entro certi limiti, fa sì che la prestazione ventricolare aumenti in parallelo con la lunghezza diastolica delle fibre. Nonostante la relazione fra queste due funzioni sia più smussata nel cuore colpito da infarto, rispetto a quello normale, essa rimane valida e tale da risultare talvolta d'importanza critica. L'aumento della pressione di riempimento, tuttavia, quando è eccessivo può favorire la congestione polmonare; è norma comune quella di non spingere la pressione diastolica ventricolare oltre i 20 mm Hg e di utilizzare la risposta clinica del paziente come la guida più sicura all'espansione di volume. Questa può essere realizzata con infusione di plasma, albumina umana, destrano o normale soluzione salina, iniziando con l'infusione relativamente rapida di 100-200 ml di liquido. Se la risposta, valutata in base al volume minuto cardiaco, pressione arteriosa e diuresi, risulta positiva, senza segni di congestione polmonare, l'infusione può essere incrementata fino alla quantità globale di 500-1.000 ml. Se l'espansione di volume provoca un aumento della pressione di riempimento del ventricolo e nessun miglioramento della sua funzione, è molto probabile che esso sia irresponsivo a questo tipo di rimedio e che un'ulteriore forzatura risulterebbe più di danno che di vantaggio. A questo punto si rende indispensabile un intervento d'ordine farmacologico specifico, meccanico o chirurgico.
a) Interventi d'ordine farmacologico.
Nello shock cardiogeno un incremento del flusso sistemico può essere ottenuto con tre tipi di rimedi farmacologici: 1) uso di vasodilatatori, a cui consegue la riduzione dell'impedenza aortica; 2) impiego di agenti ad azione inotropa positiva, da cui può derivare un incremento dello stato contrattile e una favorevole modificazione dell'emodinamica sistemica; 3) uso combinato dei due tipi di farmaci.
I vasodilatatori di impiego più comune - nitroglicerina, nitroprussiato e fentolammina - hanno effetti disparati sulla funzione miocardica per la loro diversa influenza su impedenza e preload. La nitroglicerina agisce primariamente sulle vene e la fentolammina sulle arteriole, mentre il nitroprussiato ha un effetto comparabile sui due distretti. La nitroglicerina riduce la congestione polmonare senza incrementare in modo significativo il volume minuto cardiaco. La fentolammina agisce in modo predominante sul volume minuto cardiaco. Il nitroprussiato influenza in pari misura il volume minuto e la pressione di riempimento del cuore. Fentolammina e, ancor più, nitroprussiato potenziano il loro effetto benefico se la condizione è complicata da rigurgito mitralico o da difetto settale, in quanto aumentano la frazione di sangue espulsa in aorta. Qualora gli agenti vasodilatatori risultino incapaci di riportare alla normalità la perfusione periferica, e la pressione arteriosa sia eccessivamente bassa, è appropriato ricorrere all'impiego di agenti ad azione inotropa positiva. In circostanze di marcata bradicardia è indispensabile l'uso di isoproterenolo. Norepinefrina e metaraminolo trovano impiego solo nei casi di estrema ipotensione. Va, infatti, tenuto presente che il rialzo pressorio, di per sé positivo, è in genere ottenuto a spese di una vasocostrizione sistemica, che a sua volta può tradursi in un incremento non desiderato dello sforzo dinamico del cuore e del suo consumo energetico. Fatte queste eccezioni, fra gli agenti ad azione inotropa positiva si preferiscono attualmente dopammina e dobutammina. Queste due sostanze, oltre a possedere un effetto beta-stimolante a livello miocardico, esercitano un'influenza vasodilatante diretta sui vasi renali, mesenterici e coronarici, e un'azione vasocostrittrice a livello dei muscoli scheletrici. Il risultato di questi effetti è in genere un calo della resistenza vascolare sistemica (o comunque un non incremento della medesima), un aumento non rilevante della frequenza del battito e un miglioramento del volume minuto cardiaco, che porta in genere come conseguenza l'aumento della pressione arteriosa. La vasodilatazione renale e l'incremento della diuresi sono un ulteriore elemento positivo in presenza di shock.
Talvolta può essere particolarmente utile l'uso combinato di queste sostanze con agenti vasodilatatori. In tal modo viene raggiunto un effetto sinergico che porta alla contemporanea e considerevole riduzione della congestione polmonare e a un aumento importante del flusso sistemico, entrambi in misura superiore a quella ottenibile con l'impiego separato dei due tipi di farmaci.
b) Interventi d'ordine meccanico.
Nel continuo sforzo rivolto a migliorare la prognosi dell'infarto miocardico e, in particolare, a porre rimedio ai difetti dinamici del cuore con esso relati, sussidi d'ordine meccanico si sono di recente affiancati a quelli d'ordine farmacologico. Il più diffuso fra questi, che prende il nome di contropulsazione aortica, è basato sul principio di aumentare la perfusione coronarica, ridurre il consumo energetico del cuore e dare supporto al circolo sistemico. Un palloncino della capacità di circa 30 ml, attaccato alla porzione terminale di un catetere, viene inserito nell'aorta toracica discendente attraverso un'arteria femorale. L'inflazione del palloncino in fase diastolica e la sua deflazione in fase sistolica (la sincronizzazione è fatta con l'elettrocardiogramma) fanno sì che durante la diastole la pressione aortica e la perfusione coronarica risultino aumentate, e durante la sistole risulti ridotta l'impedenza aortica, con conseguente risparmio energetico del ventricolo sinistro. I risultati immediati dell'applicazione di questa metodica sono chiaramente incoraggianti, sia sul piano biochimico (produzione di lattato), sia sul piano strettamente clinico. Un certo numero di casi di shock cardiogeno insensibile alla terapia medica mostra una risposta favorevole all'assistenza meccanica. Va detto, tuttavia, che spesso il miglioramento è di breve durata e cessa non appena viene interrotta l'assistenza meccanica. Quando ciò avviene, è presumibile che il danno miocardico sia stato grave al punto da impedire un recupero valido e permanente, nonostante l'ausilio meccanico.
c) Interventi d'ordine chirurgico.
Si tratta di interventi di emergenza e ad alto rischio, eseguiti in genere nella fase acuta dell'infarto per porre rimedio a complicanze anatomiche (quali perforazione settale, rottura di corde tendinee o di muscolo papillare) e allo shock cardiogeno non responsivo a rimedi d'ordine farmacologico e meccanico. Essi possono consistere in: resezione della zona infartuata, rivascolarizzazione con bypass coronarico, riparazione di un difetto del setto interventricolare, sostituzione di una valvola mitralica, combinazione di due o più di questi interventi.
La valutazione corretta mediante cateterismo cardiaco del tipo e della gravità del danno anatomico e la tempestività dell'intervento chirurgico sono i presupposti per il successo terapeutico. Tutto ciò richiede la stretta collaborazione e l'alta competenza di un gruppo cardiologico clinico e chirurgico, che agisca con perfetta coordinazione e abbia la pronta disponibilità delle strutture necessarie per realizzare diagnosi e terapia. Nei soggetti in cui venga evidenziata una chiara indicazione chirurgica e in cui l'intervento possa essere realizzato con successo, la sopravvivenza a distanza è intorno al 60%. Purtroppo la minoranza di coloro che sopravvivono è composta da soggetti il cui infarto è complicato da shock cardiogeno da perdita di massa contrattile, refrattario ai rimedi di tipo farmacologico.
Per concludere, nonostante gli sforzi compiuti e i progressi terapeutici più recenti, lo shock cardiogeno da infarto del miocardio rimane un problema clinico di importanza cruciale ed è tuttora un evento fatale in più di quattro quinti dei casi.
Bibliografia.
Abrams, D. L., Edelist, A., Luria, M. H., Miller, A. G., Ventricular aneurism: a reappraisal based on a study of 65 consecutive autopsied cases, in ‟Circulation", 1963, XXVII, pp. 164-169.
??? Anticoagulants in acute myocardial infarction. Results of a cooperative clinical trial, in ‟Journal of the American Medical Association", 1973, CCXXV, pp. 724-729.
Berne, R. M., Degeest, H., Levy, M. N., Influence of the cardiac nerves on coronary resistance, in ‟American journal of physiology", 1965, CCVIII, pp. 763-769.
Bjorek, G., Mogensen, L., Nyquist, O., Orinius, E., Sjogren, A., Studies of myocardial rupture with cardiac tamponade in acute myocardial infarction, in ‟Chest", 1972, LXI, pp. 4-6.
Blumenthal, M. R., Wang, H. H., Markee, S., Wang, S. G., Effects of acetylcholine on the heart, in ‟American journal of physiology", 1968, CCXIV, pp. 1280-1287.
Blumgart, H. I., Schlesinger, M. J., Zoll, P. M., Angina pectoris, coronary failure and acute myocardial infarction: role of coronary occlusions and collateral circulation, in ‟Journal of the American Medical Association", 1941, CXVI, pp. 91-99.
Bohr, D. F., Vascular smooth muscle up date, in ‟Circulation research", 1973, XXXII, pp. 665-672.
Bradley-More, P. R., Lebowitz, E., Greene, M. W., Atkins, H. L., Ansar, A. N., Thallium-201 for medical use. II. Biologic behavior, in ‟Journal of nuclear medicine", 1975, XVI, pp. 156-160.
Braunwald, E., Ross, J., Sonnenblick, E. H., Mechanisms of contraction of the normal and failing heart, Boston 19762.
Chahine, R. A., Raizner, A. E., Ishimori, T., Luchi, R. J., Mac Intosh, R. D., The incidence and clinical implications of coronary artery spasm, in ‟Circulation", 1975, LII, pp. 972-978.
Dhurandhar, R. W., Watt, D. L., Silver, M. D., Trimble, A.S., Adelman, A. G., Prinzmetal's variant form of angina with angiographic evidence of coronary arterial spasm, in ‟American journal of cardiology", 1972, XXX, pp. 902-905.
Endo, M., Hirosawa, K., Kaneko, N., Hase, K., Inoue, Y., Konno, S., Prinzmetal's variant angina. Coronary arteriogram and left ventriculogram during angina attack by metacholine, in ‟New England journal of medicine", 1976, CCXCIV, pp. 252-255.
Endo, M., Kada, I., Hosoda, S., Hayashi, H., Hirosawa, K., Konno, S., Prinzmetal's variant form of angina pectoris. Re-evaluation of mechanism, in ‟Circulation", 1975, LII, pp. 33-37.
Erhardt, L. F., Coronary thrombosis: cause or consequence, in acute and long-term medical management of myocardial ischemia (a cura di L. Withelmsen e A. Hjalinarson), Mölndal 1978, p. 41.
Fleckenstein, A., Nakayama, K., Fleckenstein-Grun, G., Byon, Y. K., Interactions of hydrogen ions, calcium antagonistic drugs and cardiac glycosides with excitation-contraction coupling of vascular smooth muscle, in Ionic action on vascular smooth muscle (a cura di E. Betz), Berlin 1976, p. 117.
Friedberg, C. K., Horn, H., Acute myocardial infarction not due to coronary artery occlusion, in ‟Journal of the American Medical Association", 1939, CXII, pp. 1675-1679.
Gaasch, W. H., Adyanthaya, A. V., Wang, V. H., Pickering, E., Quinones, M. A., Alexander, J. K., Prinzmetal's variant angina: hemodynamic and angiographic observations during pain, in ‟American journal of cardiology", 1976, XXXVII, pp. 831-839.
Gallavardin, L., Les syncopes d'effort, in ‟Lyon médical", 1973, XV, pp. 217-230.
Ganote, C. E., Worstell, J., Kaltenback, J. P., Oxygen-induced enzyme release after irreversible myocardial injury, in ‟American journal of pathology", 1976, LXXXIV, pp. 327-349.
Gellai, M., Norton, J. M., Detar, R., Evidence for direct control of coronary vascular tone by oxygen, in ‟Circulation research", 1973, XXXII, pp. 279-289.
Gold, H. K., Leinbach, R. C., Maroko, P.R., Reduction of myocardial injury in patients with acute myocardial infarction by propranolol, in ‟American journal of cardiology", 1976, XXVIII, pp. 689-695.
Gorlin, R., Pathophysiology of cardiac pain, in ‟Circulation", 1965, XXXII, pp. 138-148.
Gould, M. L., Lipscomb, K., Effects of coronary stenosis on coronary flow reserve and resistance, in ‟American journal of cardiology", 1974, XXXIV, pp. 48-55.
Guazzi, M., Magrini, F., Polese, A., Fiorentini, C., Bartorelli, C., Left and right haemodynamics during spontaneous angina pectoris. Comparison between angina with ST segment depression and angina with ST segment elevation, in ‟British heart journal", 1975, XXXVII, pp. 401-413.
Guazzi, M., Polese, A., Fiorentini, C., Magrini, F., Bartorelli, C., Left ventricular performance and related haemodynamic changes in Prinzmetal's variant angina pectoris, in ‟British heart journal", 1971, XXXIII, pp. 89-94.
Heberden, W., Some accounts of a disorder of the braest. Read before the Royal College of Physicians, July 21, 1768, in ‟Medical transactions of the Royal College of Physicians (London)", 1772, II, p. 59.
Herrick, J. B., Clinical features of sudden obstruction of the coronary arteries, in ‟Journal of the American Medical Association", 1912, LIX, pp. 2015-2020.
Higgins, C. B.,Wexler, L., Silverman, J. F., Schroeder, J. S., Clinical and arteriographic features of Prinzmetal's variant angina: documentation of etiologic factors, in ‟American journal of cardiology", 1976, XXXVII, pp. 831-839.
Hirsh, E. F., Borghard-Erdle, A. M., The innervation of the human heart, in ‟Archives of pathology", 1961, LXXI, pp. 384-407.
Isnar, J. M., Roberts, W. C., Right ventricular infarction complicating left ventricular infarction secondary to coronary heart disease. Frequency, location, associated findings and significance from analysis of 236 necropsy patients with acute or healed myocardial infarction, in ‟American journal of cardiology", 1978, XLII, pp. 884-894.
Jennings, R. B., Ganote, C. E., Structural changes in myocardium during acute ischemia, in ‟Circulation research", 1974, XLIII, suppl. 3, pp. 156-172.
Johnson, A. D., Detwiler, J. H., Coronary spasm, variant angina and recurrent myocardial infarctions, in ‟Circulation", 1977, LV, pp. 947-950.
King, M. J., Zir, L. M., Kaltman, A. J., Fox, A. C., Variant angina associated with angiographically demonstrated coronary artery spasm and REM sleep, in ‟American journal of the medical sciences", 1973, CCLXV, pp. 419-422.
Klein, M. S., Shell, W. E., Sobel, B. E., Serum creatine-phosphokinase (CPK) isoenzymes following intramuscular injection, surgery and myocardial infarction. Experimental and clinical studies, in ‟Cardiovascular research", 1973, VII, pp. 412-418.
Kloner, R. A., Ganote, C. E., Whalen, D. A., Sennings, R. B., Effect of transient period of ischemia on myocardial cells. II. Fine structure during the first few minutes of reflow, in ‟American journal of pathology", 1974, LXXIV, pp. 399-413.
LaDue, J. S., Wrahlewski, F., Karnien, A., Serum glutamic oxalacetic transaminase activity in human acute transmural myocardial infarction, in ‟Science", 1954, CXX, pp. 497-499.
Lange, R. L., Reid, M. S., Tresch, D.D., Keelan, M. H., Bernhard, V. M., Coolidge, G., Nonatheromatous ischemic heart disease following withdrawal from chronic industrial nitroglycerin exposure, in ‟Circulation", 1972, XLVI, pp. 666-678.
Latham, P. M., Collected works, London 1876.
Levy, M. N., Sympathetic-parasympathethic interaction in the heart, in ‟Circulation research", 1971, XXIX, pp. 437-445.
Linhart, J. W., Prinzmetal's variant of angina pectoris, in ‟Journal of the American Medical Association", 1974, CCXXVIII, pp. 342-343.
MacAlpin, R. N., Coronary spasm as a cause of angina, in ‟New England journal of medicine", 1973, CCLXXXVIII, pp. 788-789.
MacAlpin, R. N., Kattus, A. A., Alvaro, A. B., Angina pectoris at rest with preservation of exercise capacity. Prinzmetal's variant angina, in ‟Circulation", 1973, XLVII, pp. 946-958.
Malliani, A., Schwartz, P. J., Zanchetti, A., A sympathetic reflex elicited by experimental coronary occlusion, in ‟American journal of physiology", 1969, CCXVII, pp. 703-709.
Maseri, A., Mimmo, R., Chierchia, S., Marchesi, C., Pesola, A., L'Abbate, A., Coronary artery spasm as a cause of acute myocardial ischemia in man, in ‟Chest", 1975, LXVIII, pp. 625-633.
Meller, J., Pichard, A., Dack, S., Coronary arterial spasm in Prinzmetal's angina: a proved hypotesis, in ‟American journal of cardiology", 1976, XXXVII, pp. 938-940.
Modan, B., Shani, M., Schor, S., Reducation of hospital mortality from acute myocardial infarction by anticoagulant therapy, in ‟New England journal of medicine", 1975, CCXCII, pp. 1359-1362.
Mrwa, U., Achtig, I., Rüegg, J. C., Influences of calcium concentration and pH on the tension development and ATP-ase activity on the arterial actomyosin contractile system, in ‟Blood vessels", 1974, XI, pp. 277-286.
Mueller, H. S., Ayres, S. M., Religa, A., Evans, A. G., Propranolol in the treatment of acute myocardial infarction. Effect on myocardial oxygenation and hemodynamics, in ‟Circulation", 1974, XLIX, pp. 1078-1087.
Nayler, W., Crau, A., Slade, A., A protective effect of verapamil on hypoxic heart muscle, in ‟Cardiovascular research", 1976, X, pp. 650-662.
Oliva, P. B., Breckinridge, J. C., Arteriographic evidence of coronary arterial spasm in acute myocardial infarction, in ‟Circulation", 1977, LVI, pp. 366-374.
Oliva, P. B., Potts, D. E., Pluss, R. G., Coronary arterial spasm in Prinzmetal's angina. Documentation by coronary arteriography, in ‟New England journal of medicine", 1973, CCLXXXVIII, pp. 745-751.
Olson, H. G., Lysons, K. P., Aronow, W.S., Brown, T., Greenfield, R. S., Follow-up technetium-99m stannous pyrophosphate myocardial scintigrams after acute myocardial infarction, in ‟Circulation", 1977, XXXVI, pp. 801-815.
Osler, W., Lumleian lectures on angina pectoris, in ‟Lancet", 1910, I, pp. 697-699.
Peter, C. T., Norris, R. M., Clarke, E. D., Heng, M. K., Singh, B. N., Williams, B., Howell, D. R., Ambler, P. K., Reduction of enzyme release after acute myocardial infarction by propranolol, in ‟Circulation", 1978, LVII, pp. 1091-1095.
Prinzmetal, M., Kennamer, R., Merlis, R., Wada, T., Bor, N., A variant form of angina pectoris: preliminary report, in ‟American journal of medicine", 1959, XXVII, pp. 375-388.
Prinzmetal, M., Schwartz, L. L., Carday, E., Spritzler, R., Bergman, H. C., Kruger, H. E., Studies on the coronary circulation. VI. Loss of myocardial contractility after coronary artery occlusion, in ‟Annals of internal medicine", 1949, XXI, pp. 429-449.
Reimer, K. A., Lowe, J. E., Jenning, R. B., Effect of the calcium antagonist verapamil on necrosis following temporary coronary artery occlusion in dogs, in ‟Circulation", 1977, LV, pp. 581-587.
Riley, C. P., Russel, R. O. Jr., Rackley, C. E., Left ventricular gallop sound and acute myocardial infarction, in ‟American heart journal", 1973, LXXXVI, pp. 598-602.
Roberts, W.C., Coronary arteries in fatal acute myocardial infarction, in ‟Circulation", 1972, XLV, pp. 215-230.
Ross, R. S., Gorlin, R., Coronary arteriography, in ‟Circulation", 1968, XXXVIII, pp. 67-73.
Rüegg, J. C., Smooth muscle tone, in ‟Physiological reviews", 1971, LI, pp. 201-248.
Samson, W. E., Scher, A. M., Mechanism of ST segment alteration during acute myocardial injury, in ‟Circulation research", 1964, VIII, pp. 780-787.
Selzer, A., Cardiac ischemic pain in patients with normal coronary arteriograms, in ‟American journal of medicine", 1977, LXIII, pp. 661-665.
Strauss, H. W., Harrison, K., Langan, J. K., Lebowitz, E., Pitt, B., Thallium-201 for myocardial imaging, in ‟Circulation", 1975, LI, pp. 641-645.
??? Sulfinpyrazone in the prevention of cardiac death after myocardial infarction. The anturane reinfarction study, in ‟New England journal of medicine", 1978, CCXCVIII, pp. 289-295.
Tennant, R., Wiggers, C. J., Effect of coronary occlusion on myocardial contraction, in ‟American journal of physiology", 1935, CXII, pp. 351-361.
Thoren, P. N., Activation of the left ventricular receptors with nonmedullated vagal afferent fibers during occlusion of a coronary artery in the cat, in ‟American journal of cardiology", 1976, XXXVII, pp. 1046-1051.
Tonascia, J., Gordin, L., Schmerler, H., Retrospective evidence favoring use of anticoagulants for myocardial infarction, in ‟New England journal of medicine", 1975, CCXCII, pp. 1362-1366.
Vlodaver, Z., Coe, J. L., Edwards, J. E., True and false left ventricular aneurism, in ‟Circulation", 1975, LI, pp. 567-572.
Waagstein, F., Hjamalrson, A.C., Double-blind study of the effect of cardioselective beta-blockade on chest pain in acute myocardial infarction, in ‟Acta medica scandinavica", 1976, DLXXXVII, suppl., pp. 201-207.
Wade, W. G., The pathogenesis of infarction of the right ventricle, in ‟British heart journal", 1957, XXI, pp. 545---554.
Wartman, W. B., Hellerstein, H. K., The incidence of heart disease in 2.000 consecutive autopsies, in ‟Annals of internal medicine", 1948, XXVIII, pp. 41-45.
Wei, J. Y., Hutchins, G. M., Bulkley, B. H., Papillary muscle rupture in fatal acute myocardial infarction, in ‟Annals of internal medicine", 1979, XC, pp. 149-152.
Wierner, L., Kasparian, H., Duca, P. R., Walinsky, P., Gottlieb, R. S., Hanckel, F., Brest, A. N., Spectrum of coronary arterial spasm. Clinical, angiographic and myocardial metabolic experience in 29 cases, in ‟American journal of cardiology", 1976, XXXVIII, pp. 945-955.
Wright, I.S., Marple, C.D., Beck, D. F., Report of the committee for the evaluation of anticoagulants in the treatment of coronary thrombosis with myocardial infarction, in ‟American heart journal", 1948, XXXVI, pp. 801-815.
Yasue, H., Nagao, M., Omote, S., Takizawa, A., Miwa, K., Tanaka, S., Coronary arterial spasm and Prinzmetal's variant form of angina induced by hyperventilation and tris-buffer infusion, in ‟Circulation", 1978, LVIII, pp. 56-62.
Yasue, H., Touyama, M., Kato, H., Tanaka, S., Akiyama, F., Prinzmetal's variant form of angina as a manifestation of alpha-adrenergic receptor-mediated coronary artery spasm: documentation by coronary arteriography, in ‟American heart journal", 1976, XCI, pp. 148-155.
Yasue, H., Touyama, M., Shimamoto, M., Hirafumi, K., Tanaka, S., Akiyama, H., Role of autonomic nervous system in the pathogenesis of Prinzmetal's variant form of angina, in ‟Circulation", 1974, L, pp. 534-539.
Zuberbuhler, R.C., Bohr, D. F., Responses of coronary smooth muscle to catecholamines, in ‟Circulation research", 1965, XVI, pp. 431-440.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


