Reazioni chimiche
Enciclopedia della Scienza e della Tecnica (2007)
Reazioni chimiche
I problemi della chimica gravitano intorno a due argomenti di fondo, che riguardano rispettivamente la struttura delle molecole e le loro trasformazioni. Infatti, le molecole, a seconda della loro natura e delle condizioni in cui si trovano, possono subire modifiche che sono generalmente associate a scambi di energia con le altre molecole o, più in generale, con l'ambiente circostante. Ogni trasformazione che comporti la rottura e la formazione di legami chimici, la cui energia è dell'ordine di 50 kcal/mole (o più) costituisce una reazione chimica. A ognuna di esse corrisponde un'equazione stechiometrica che, rispecchiando la legge della conservazione della massa e degli elementi, contiene la prima essenziale informazione sperimentale di cui si deve tener conto per ogni ulteriore approfondimento. Per esempio, per la formazione di ammoniaca 3H2+N2 = 2NH3 l'equazione stechiometrica registra il fatto sperimentale che due moli di ammoniaca si formano da tre moli di idrogeno e una di azoto.
Ma l'equazione stechiometrica è ben lungi dall'esaurire la descrizione delle caratteristiche di una reazione chimica perché lascia aperti due quesiti fondamentali: perché avviene e come avviene una trasformazione chimica.
Una reazione chimica avviene spontaneamente solo a condizione che essa comporti una diminuzione dell'energia libera di Gibbs G del sistema materiale che vi prende parte. La grandezza G si compone di due parti ‒ l'entalpia H e l'entropia S ‒ legate fra loro dall'equazione
[1] ΔG = ΔH − TΔS.
Sebbene il contributo del termine entropico, TΔS, sia spesso modesto, per cui la reazione risulta dominata da una diminuzione dell'entalpia (reazioni esotermiche), esistono casi in cui il termine entropico è determinante perché la reazione avvenga spontaneamente. È comunque essenziale che si abbia ΔG 〈 0, qualunque sia la combinazione algebrica dei due termini entalpico ed entropico. Le reazioni, quindi, avvengono perché sono associate a una perdita di energia potenziale, che, al contrario, deve essere fornita ove si voglia far retrocedere la reazione stessa.
Anche se una reazione può avvenire in modo spontaneo, debbono essere soddisfatte altre condizioni perché essa proceda entro un tempo sufficientemente breve da essere osservabile. Una miscela di metano e ossigeno, per esempio, si conserva per un tempo praticamente indeterminato a temperatura ambiente, anche se la combustione dell'idrocarburo va nel senso della reazione spontanea CH4 + 2O2 → CO2 + 2H2O.

Tali condizioni coinvolgono i dettagli con cui la reazione avviene e che definiscono il meccanismo di reazione. Pertanto il tempo di reazione è una variabile essenziale delle trasformazioni chimiche ed è la spia del meccanismo di reazione. Nel corso di una reazione chimica ha luogo una ridistribuzione degli atomi di una o più specie molecolari mediante la rottura e la formazione di legami chimici. (fig. 1)
Sommario
1. Come avviene una reazione chimica. 2. Metodi di studio. 3. Reazioni dei composti del carbonio . 4. Reazioni dei composti degli elementi diversi dal carbonio. 5. Considerazioni e prospettive sullo stato delle conoscenze delle reazioni chimiche. □ Bibliografia.
Come avviene una reazione chimica
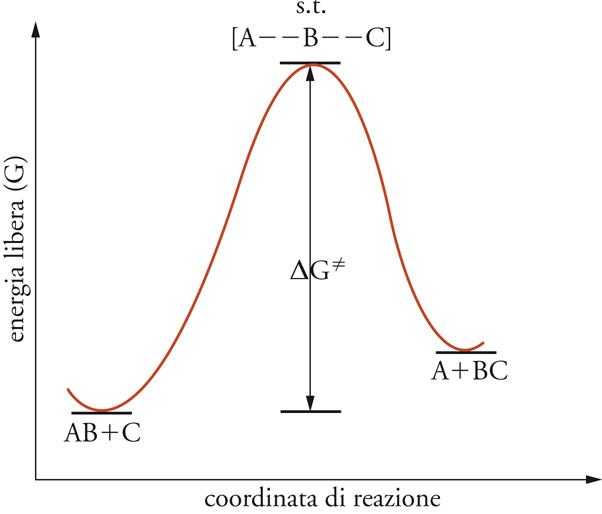
Una reazione chimica avviene mediante il movimento delle molecole, il loro incontro, la rottura dei loro legami, il trasferimento di frammenti molecolari e il comporsi di nuove specie molecolari. È nella particolare sequenza di questi eventi che il sistema trova il percorso energeticamente più favorevole affinché la trasformazione abbia luogo. In ogni caso, occorre trasferire energia perché la reazione avvenga. Secondo la teoria delle velocità assolute di reazione, il sistema dei reagenti si trasforma nei prodotti passando attraverso una speciale condizione critica detta stato di transizione o complesso attivato, nella quale l'energia libera del sistema raggiunge un massimo (fig. 2).
Solitamente il complesso attivato, indicato con il simbolo ≠, non può essere considerato un vero composto chimico, poiché ha una vita media comparabile ai periodi di vibrazione dei legami chimici stessi. La differenza di energia libera tra complesso attivato e sistema dei reagenti, detta energia libera di attivazione, indicata con il simbolo ΔG≠, fornisce una misura della reattività chimica. Quanto più alta è tale differenza tanto più lenta è la reazione. La teoria prevede che anche ΔG≠ abbia una componente entalpica e una entropica correlabili mediante l'equazione
[2] ΔG≠ = ΔH≠ − TΔS≠
nella quale i parametri di attivazione indicano le differenze tra lo stato di transizione e i reagenti.
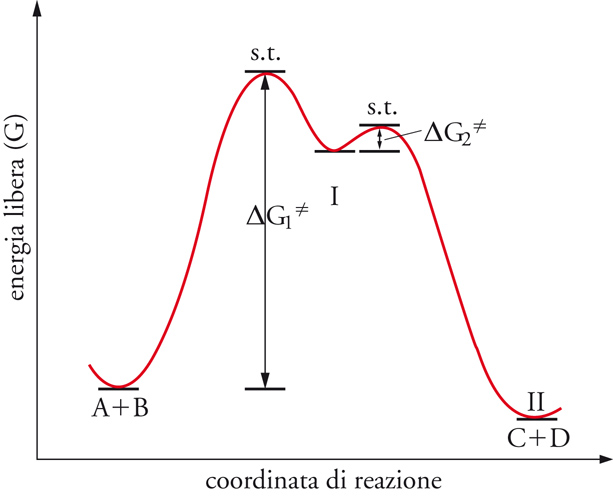
Il caso illustrato nella fig. 2 è quello di una trasformazione a un solo stadio. Quando la reazione avviene a più stadi, esiste uno stato di transizione per ciascuno di essi. Il passaggio da uno stadio all'altro è caratterizzato da un prodotto intermedio cui corrisponde un minimo nel diagramma dell'energia libera (fig. 3).
Le condizioni sperimentali ‒ quali la natura dei reagenti, del solvente e di altre sostanze aggiunte ‒ possono avere effetti notevoli sull'energia libera di attivazione, così da provocare abbassamenti di ΔG≠ fino a varie kcal/mole, che corrispondono a notevoli incrementi della velocità di reazione. Lo studio della velocità di reazione, ossia della cinetica chimica, è alla base dello studio dei meccanismi di reazione, la cui conoscenza comporta l'individuazione degli eventuali prodotti intermedi.
Le caratteristiche generali dei meccanismi di reazione sono note per molte reazioni chimiche, ma spesso la loro comprensione dettagliata è un'impresa molto ardua. Le nostre conoscenze in questo campo vengono continuamente aggiornate man mano che vengono raccolti nuovi dati sperimentali. Un meccanismo di reazione attendibile deve poter spiegare tutti i fatti sperimentali noti, che talora, però, sono compatibili con più varianti diverse.
L'obiettivo di questi studi non è solo teorico, perché si propone di individuare regole generali che consentano di guidare il chimico alla ricerca di nuove reazioni, di migliorare i metodi di sintesi, di produrre prodotti industriali a minor costo o con migliori prestazioni, di approfondire le conoscenze dei meccanismi chimici fondamentali che sottostanno ai processi biologici.
Metodi di studio
Per quanto riguarda la rottura e la formazione dei legami chimici, le molecole si comportano nei modi seguenti:
(a) un legame si rompe in modo che entrambi gli elettroni passano a uno dei frammenti risultanti,
[3] A:B → A: + B;
(b) un legame si forma in modo che i due elettroni provengono da uno stesso atomo,
[4] A: + C → A:C
in questo caso si ha una scissione eterolitica o ionica, perché spesso si formano intermedi ionici.
Se la trasformazione è il risultato dell'interazione tra due diverse sostanze, prende il nome di substrato quella di cui si intende studiare il comportamento e di reagente quella con cui il substrato reagisce. Se il reagente interviene nella formazione dei legami chimici, come per esempio A nella [4], mettendo a disposizione i due elettroni, esso prende il nome di reagente nucleofilo; viceversa, se accetta gli elettroni, come per esempio C, si chiama reagente elettrofilo.
Se nella rottura dei legami, a ciascun frammento risultante si associa un elettrone
[5] A:B → A∙ + B∙
si generano radicali liberi, ossia specie fornite di elettroni non condivisi. In questo caso si hanno reazioni omolitiche o radicaliche.
Ciò premesso, ricordiamo che molte reazioni avvengono in due o più stadi successivi e passano attraverso la formazione di prodotti intermedi che, essendo spesso molto labili, subiscono ulteriori trasformazioni verso i prodotti finali della reazione.
Una parte importante dello studio consiste nello stabilire se e quali prodotti intermedi si formano durante la reazione. Può accadere che l'intermedio si accumuli in una certa misura nel corso della reazione e prima che questa abbia termine. Se s'interrompe la reazione e/o si cerca di farla avvenire in condizioni più moderate, si può favorire tale accumulo al punto da poterlo isolare, ovvero da poterlo intercettare nella miscela nella quale si forma attraverso qualche sua proprietà spettrale (spettri UV, visibile, IR, NMR, ecc.). Se si tratta questo composto nelle stesse condizioni sperimentali, si ottengono gli stessi prodotti finali di reazione in un tempo non superiore a quello richiesto dalla reazione in esame.
Informazioni univoche sugli atomi particolari che prendono parte a una reazione si possono ottenere marcando le molecole con isotopi. Per esempio, dall'analisi dei prodotti ottenuti nella trasposizione di Claisen, non è possibile dedurre se nella migrazione del gruppo allilico si lega all'anello aromatico il carbonio-a o il carbonio-c.
[6] formula
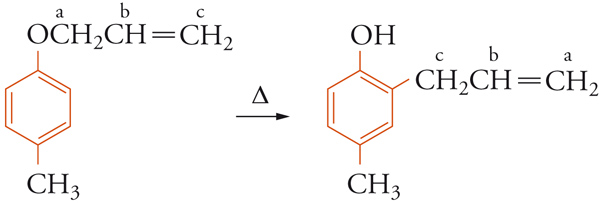
Ma se si parte da un etere allilico marcato nel carbonio-c con l'isotopo radioattivo 14C e si ricerca la posizione di tale atomo nel prodotto con misure di radioattività su opportuni prodotti di decomposizione, si trova che nella migrazione della catena allilica è il carbonio-c che forma il legame con il carbonio aromatico in posizione orto rispetto all'ossigeno.
Un metodo molto efficace per lo studio delle reazioni chimiche è quello stereochimico, che consiste nel seguire le eventuali variazioni di configurazione degli atomi nel corso della reazione. Per esempio, se un dato atomo di carbonio del substrato è asimmetrico ed è sede di una trasformazione chimica, nel prodotto si può ritrovare con la configurazione invertita, racemizzata o conservata. Questi risultati hanno implicazioni significative sul percorso stereochimico della reazione e, in particolare, sulla direzione di attacco del reagente e sulla configurazione del complesso attivato. Essi si sono rivelati di grande utilità nello studio di molte reazioni organiche ed elemento-organiche.
Accanto a tali analisi è importante esaminare il comportamento cinetico del sistema reagente il quale fornisce uno strumento essenziale per lo studio delle reazioni chimiche. Lo studio cinetico consiste nel determinare la velocità con la quale avviene la reazione a una o più temperature e nell'individuarne la dipendenza dalle variabili sperimentali (concentrazione, solvente, struttura, catalizzatori, ecc.).
La velocità di una reazione chimica, che solitamente viene indicata con r, esprime la quantità di materia che si trasforma nell'unità di tempo e di volume del sistema reagente. Un'importante legge che regola le trasformazioni più semplici, detta legge di azione di massa, stabilisce che la velocità di reazione è proporzionale al prodotto delle concentrazioni delle sostanze che prendono parte alla reazione. Per esempio, se la sostanza S subisce una trasformazione unimolecolare e si trasforma in P, ne risulta una velocità r che segue l'equazione del 1° ordine
[7] r = kCS
dove k è la costante specifica di velocità e CS la concentrazione molare del reagente. Se avviene una trasformazione bimolecolare della sostanza S o una reazione tra due sostanze S′ e S″, si ottiene un'equazione cinetica del 2° ordine
[8] r = kCS2
ovvero
[9] r = kCS′CS″ .
La molecolarità, invece, indica il numero delle molecole che partecipano alla formazione del complesso attivato ed è pertanto un aspetto molto importante del meccanismo di reazione.
In realtà, molte reazioni chimiche non coinvolgono trasformazioni semplici perché decorrono attraverso una successione di più stadi. In tali casi l'ordine cinetico (determinato sperimentalmente e riferito all'intero processo) non fornisce indicazioni sulle molecolarità della reazione, che vanno stabilite stadio per stadio. La complessità del meccanismo di reazione si può dedurre da eventi diversi: per esempio, la velocità può non dipendere dalla concentrazione di una delle sostanze reagenti; ovvero può dipendere dalla concentrazione di una sostanza che non appare come reagente nell'equazione chimica della reazione in esame. Questi fatti indicano che la reazione procede in più stadi. Il meccanismo della reazione è definito dalla sequenza dei vari stadi e dalla natura dei prodotti intermedi che si formano tra uno stadio e l'altro. Quello più lento condiziona la velocità dell'intero processo, qualunque sia la sua posizione nella sequenza.
Per determinare la velocità di reazione si deve disporre di un metodo accurato di analisi per misurare la concentrazione di uno o più componenti della miscela di reazione. I metodi di analisi variano da caso a caso, ma spesso si ricorre alla variazione dello spettro di assorbimento UV o di quello visibile che accompagna la scomparsa delle sostanze di partenza e la formazione dei prodotti. Possono essere impiegati anche altri metodi spettrali (IR, NMR, ESR), come pure altre tecniche analitiche, quali l'analisi volumetrica, l'analisi potenziometrica e così via. Se l'analisi richiede il prelevamento di campioni, la reazione in ciascun campione va fermata al tempo prescelto, aggiungendo un opportuno reagente oppure con un altro metodo. Talvolta si ricorre a metodi di analisi non chimici, quali quelli che misurano la variazione di volume (dilatometria) della miscela, la variazione di pressione (manometria), se si tratta di reazioni in fase gassosa, lo svolgimento di calore (calorimetria).
Lo studio dell'influenza della temperatura sulle costanti di velocità conduce alla determinazione delle entalpie e delle entropie di attivazione, i cui contributi all'energia libera di attivazione gettano luce, per esempio, sull'importanza relativa degli effetti legati alla rottura (e alla formazione) dei legami chimici e degli effetti di repulsione sterica ΔH≠ e di solvatazione ΔS≠.
Lo studio delle proprietà del solvente e dei suoi effetti cinetici, che è stato sviluppato con grande interesse, ha permesso di individuare varie categorie di solventi e di chiarire la natura delle interazioni soluto-solvente sia nello stato fondamentale sia nello stato di transizione dei sistemi reagenti. In tal modo l'effetto del solvente può fornire un importante criterio diagnostico del meccanismo.
Un notevole grado di approfondimento su particolari aspetti di una reazione, si può raggiungere attraverso lo studio dell'effetto che provoca sulla velocità di reazione la sostituzione di un determinato atomo con un suo isotopo. La teoria degli effetti isotopici cinetici ha permesso di ottenere risultati interessanti, specialmente nel caso degli isotopi dell'idrogeno, usati molto frequentemente per stabilire, per esempio, se e in che misura la rottura del legame C-H sia avvenuta nello stadio lento di un processo.
Nelle reazioni chimiche che saranno illustrate in seguito si vedrà che uno dei problemi chiave è quello di dimostrare se il sistema in reazione passa attraverso intermedi che differiscono dal centro di reazione iniziale (atomo di carbonio o altro atomo) per un numero di coordinazione inferiore o superiore. Il primo caso si verifica in seguito alla dissociazione di un legante
[10] MXn → MXn−1+ X;
il secondo quando un reagente Y si addiziona al substrato legandosi al centro di reazione
[11] MXn + Y → MXnY.
Questi intermedi ricorrono frequentemente nelle reazioni chimiche, ma per la loro elevata reattività se ne può dimostrare la presenza con metodi generalmente indiretti: cinetici, stereochimici e spettroscopici. Solo in condizioni particolari è stato possibile isolarli. Gli intermedi maggiormente studiati sono quelli che derivano dal carbonio tetraedrico per rottura di un legame chimico e, pertanto, contengono un carbonio tricoordinato. Quelli che si formano per eterolisi sono specie ioniche positive (carbocationi), per esempio CH3+, o negative (carbanioni), per esempio C6H5CH2−. In seguito a omolisi, invece, si formano radicali liberi, per esempio CH3∙. I carbocationi sono resi più stabili dai gruppi alchilici o da quelli arilici, in grado, con meccanismi diversi di trasmissione elettronica, di disperdere in qualche misura la carica del centro positivo rilasciando elettroni verso di esso. Così il catione (CH3)3 C+ (terbutilico) è molto più stabile del catione CH3+. Al contrario, i carbanioni sono stabilizzati da gruppi che disperdono la carica del centro negativo attraendola verso di sé. L'anione −CH2NO2 è molto più stabile di −CH2C6H5 per il forte effetto di attrazione elettronica del nitrogruppo.
Reazioni dei composti del carbonio
Sebbene la chimica dei composti del carbonio sia molto vasta, le corrispondenti reazioni si possono ricondurre a una tipologia limitata: le sostituzioni, le addizioni, le eliminazioni e le trasposizioni. A queste si aggiungono le polimerizzazioni, le ossidazioni e le riduzioni, che si possono anche far rientrare nei tipi precedenti o in combinazioni di essi. I primi tre tipi sono schematicamente definiti qui di seguito. Le trasposizioni danno luogo, caratteristicamente, a cambiamenti nello scheletro molecolare del substrato, mentre le polimerizzazioni permettono lo sviluppo di strutture macromolecolari mediante la ripetizione indefinita di un processo; infine, le ossidoriduzioni trasferiscono elettroni fra reagente e substrato.
Sostituzioni nucleofile al carbonio saturo
Numerose classi di derivati alchilici subiscono la sostituzione eterolitica del gruppo X da parte di un reagente nucleofilo Y−, secondo la seguente equazione:
[12] R−X + Y− → R−Y + X−
con cui si intende che nel derivato alchilico, RX, la reazione abbia luogo in corrispondenza di un atomo di carbonio tetraedrico (o saturo) al quale è inizialmente legato il gruppo uscente X, quindi sostituito da Y. Queste reazioni comprendono un gran numero di substrati diversi, tra cui gli alogenuri, gli esteri di acidi solfonici, gli esteri di acidi carbossilici, gli alcoli, gli eteri, gli ioni solfonio, le ammine, gli ioni ammonio. Per reazione con una grande varietà di reagenti nucleofili (Cl−, OH−, RO−, RS−, CN−, ROH, RNH2, ecc.) si formano alogenuri, alcoli, eteri, solfuri, nitrili, ammine e così via. Poiché molte di queste reazioni hanno importanza come metodi di sintesi, sono tra le reazioni organiche meglio studiate dal punto di vista del meccanismo.
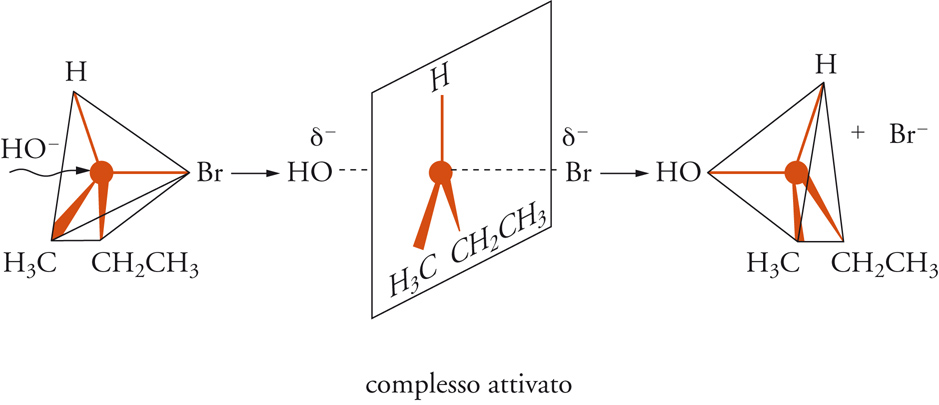
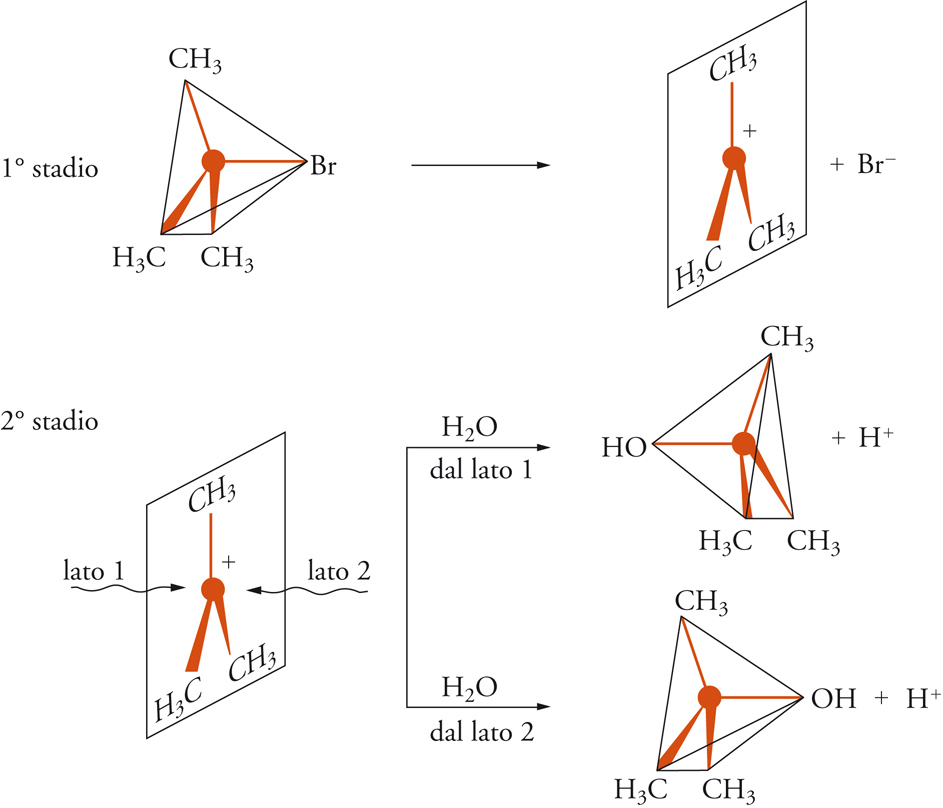
Qualsiasi trasformazione corrispondente all'equazione precedente comprende la rottura di un legame covalente (C−X) e la formazione di un nuovo legame (C−Y). Il meccanismo di questo tipo di reazione è sensibile alla struttura del substrato e alle condizioni sperimentali alle quali esso è sottoposto. Christopher K. Ingold e alcuni suoi collaboratori, proposero due meccanismi designati con i simboli SN2 e SN1, che si riferiscono, rispettivamente, alle sostituzioni nucleofile bimolecolari e monomolecolari, i cui modelli tipici sono illustrati nella fig. 4 e nella fig. 5, rispettivamente.
Il meccanismo SN2 è un processo bimolecolare e concertato, nel quale le molecole reagenti del derivato alchilico e del nucleofilo, si trasformano nei prodotti finali in uno stadio unico, secondo lo schema seguente:
[13] R − X + Y− → [Y ... R ... X]≠ → RY + X−.
Il processo è bimolecolare, perché entrambe le molecole dei reagenti fanno parte del complesso attivato nello stato di transizione, ed è concertato, perché la formazione del legame C−Y e la rottura del legame C−X avvengono simultaneamente. Il meccanismo SN1, invece, consiste in un processo a due stadi, come indicato nel seguente schema:
lento
R − X —→ Rδ+ ... Xδ− → R+ + X−
[14]
veloce
R+ + Y− —→ R − Y.
Nel primo stadio avviene la ionizzazione del legame C−X con conseguente formazione di un carbocatione come intermedio altamente reattivo. Nel secondo stadio il carbocatione reagisce con il nucleofilo trasformandosi nel prodotto finale. Lo stadio chiave di questo meccanismo è la ionizzazione lenta, cui corrisponde un complesso attivato comprendente soltanto una delle molecole reagenti; da questo punto di vista tale meccanismo si può considerare unimolecolare.
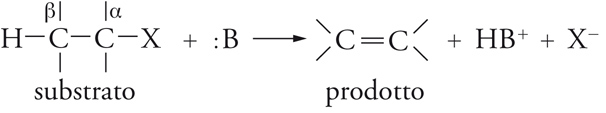
Reazioni di β-eliminazione
Queste reazioni possono essere rappresentate mediante uno schema generale che consiste nell'eliminazione del gruppo uscente X insieme a un protone dalla posizione β in presenza di una base :B. Esse ‒ come le deidroalogenazioni (da alogenuri alchilici) e le disidratazioni (da alcoli) ‒ sono importanti nelle sintesi, perché servono alla preparazione delle olefine:
[15] formula
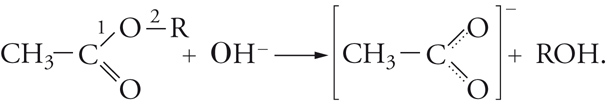
Idrolisi basica degli esteri
La saponificazione dei grassi e l'idrolisi delle ammidi fanno parte di un importante gruppo di reazioni degli acidi carbossilici e dei loro derivati di formula generale RCOX, che consistono nella sostituzione del gruppo X con un reagente nucleofilo Y−. Esso, inoltre, si ricollega, per citarne solo alcune, alle reazioni dei legami peptidici delle sostanze proteiche, alla condensazione di Claisen e alla condensazione di Dieckmann, anch'esse di notevole interesse nelle sintesi:
[16] R − COX + Y− → R − COY + X−.
Tra queste vi è l'idrolisi basica degli esteri che offre interessanti esempi di metodi di studio delle reazioni chimiche:
[17] formula
Vi sono vari modi di idrolisi degli esteri, due dei quali (alternativi) derivano dal punto di rottura dell'ossigeno del gruppo OR, la quale può avvenire in 1 o in 2, cioè, rispettivamente, nel legame acile-ossigeno o nel legame alchile-ossigeno. I numerosi studi su questa reazione portano a concludere che, negli esteri più semplici, il meccanismo di reazione comporta i seguenti stadi:
[18] formula[19]
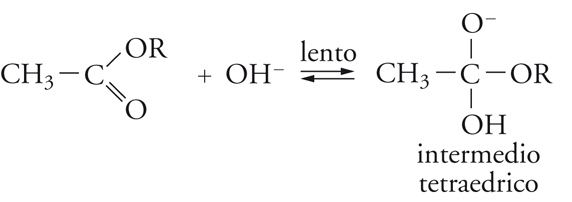
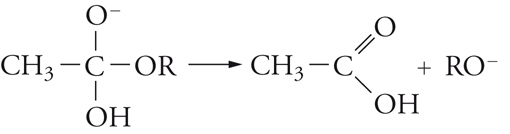
[20] formula
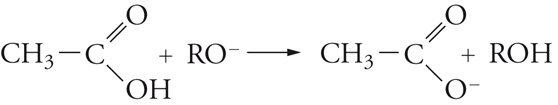
La sostituzione, quindi, avviene attraverso un'addizione che dà luogo a un intermedio in cui il carbonio acilico diventa tetraedrico, seguita da un'eliminazione dell'anione RO−, quindi, dalla dissociazione dell'acido carbossilico formatosi.
Addizioni elettrofile
Tra le reazioni dei reagenti elettrofili va ricordata l'addizione al doppio legame C=C:
[21] formula

Il doppio legame degli alcheni ha una particolare tendenza a reagire con questo tipo di reagenti, perché il legame π, relativamente debole (paragonato al legame σ si comporta come donatore di elettroni e, pertanto, è vulnerabile all'azione degli accettori). Molte sono le reazioni di questo gruppo: addizioni di acidi, di acqua, di alcoli, di alogeni, di acidi ipoalogenosi, di perossidi e così via. Di particolare interesse sono le polimerizzazioni ioniche, perché impiegate nei processi industriali per la sintesi di alti polimeri.
Reazioni radicaliche
Nelle reazioni radicaliche rientrano i tipi fondamentali di trasformazioni di tutte le reazioni organiche: sostituzione, addizione, eliminazione e trasposizione. La natura particolare di tali reazioni può essere diagnosticata dalle condizioni sperimentali nelle quali avvengono. Generalmente, esse sono provocate dalla luce o dal riscaldamento e catalizzate da alcuni perossidi. Sono insensibili all'azione degli acidi e delle basi, e possono avvenire ugualmente bene sia in fase gassosa sia in soluzione.
Infine, le reazioni radicaliche sono rallentate o soppresse da sostanze, dette inibitori, in grado di catturare rapidamente i radicali liberi che si formano e, pertanto, di bloccare un processo radicalico: gli inibitori più importanti sono l'ossigeno molecolare e il benzochinone.
Reazioni dei composti degli elementi diversi dal carbonio
Il grande sviluppo delle nostre conoscenze sulle reazioni dei composti del carbonio (reazioni organiche) ha contribuito in modo notevole a fornire i criteri e le metodologie per lo studio della dinamica chimica in generale ed è servito di stimolo alle ricerche sui meccanismi di reazione dei composti degli elementi diversi dal carbonio (reazioni inorganiche e metallorganiche). Il campo di indagine si è arricchito di nuovi orizzonti offerti dalla grande varietà delle strutture elettroniche degli elementi del sistema periodico e dalla più elevata complessità della maggior parte di essi rispetto al carbonio e agli altri elementi leggeri del Periodo 2. Le conoscenze si sono estese praticamente a tutti i membri del sistema periodico, sia agli elementi di transizione sia a quelli non di transizione.
Si deve subito notare che, nonostante la loro grande diversità, le reazioni chimiche si possono inquadrare in un numero ristretto di tipi fondamentali, anche se ciascuno di essi sottintende una grandissima varietà di comportamenti che dipendono dalla natura delle molecole specifiche e dalle condizioni sperimentali.
Dal punto di vista dei metodi di studio, valgono sostanzialmente quelli già incontrati per le reazioni del carbonio, cioè i metodi cinetici, stereochimici e isotopici. Inoltre, nelle reazioni non organiche sono molto frequenti i casi di processi estremamente rapidi che richiedono tecniche di flusso, rilassamento e risonanza magnetica nucleare o NMR (Nuclear magnetic resonance). Una spinta determinante a questi studi è stata data dallo svilupparsi di queste tecniche. La risonanza magnetica nucleare si è rivelata di grande importanza per lo studio degli aspetti stereochimici, tra cui il destino delle configurazioni enantiomeriche e la pseudorotazione.
Anche nelle reazioni non organiche si possono distinguere i due grandi gruppi di processi ‒ eterolitici e omolitici ‒ già visti per quelle organiche, e dipendenti dal modo in cui si rompono e si formano i legami di coordinazione intorno a un dato atomo. Un processo omolitico che coinvolge un atomo metallico conduce generalmente a un'ossidoriduzione. Come già detto, i reagenti possono essere nucleofili, elettrofili e radicalici.
Nella formulazione più generale della trasformazione che subisce un composto inorganico, è utile considerare la molecola come derivante da un atomo M coordinato da un certo numero di leganti L, ossia MLx. In tal caso si possono distinguere: (a) reazioni che riguardano la sfera di coordinazione di M; (b) reazioni che interessano lo stato di ossidazione di M; (c) reazioni che riguardano i leganti.
Queste ultime sono di grande interesse perché si ritrovano in molti processi catalitici importanti per sintesi di interesse industriale, nei quali i leganti sono organici. Tra le reazioni del gruppo (a) possiamo riconoscere tipi di trasformazioni corrispondenti a quelli incontrati tra le reazioni organiche: sostituzione di legante, aumento del numero di coordinazione (addizione), diminuzione del numero di coordinazione (eliminazione), trasposizione di leganti. Inoltre, le molecole MLx possono subire modifiche di configurazione.
Il numero di coordinazione e la configurazione condizionano notevolmente il modo di reagire delle molecole. Pertanto forniscono un criterio di suddivisione delle reazioni dei composti inorganici. I principali gruppi di molecole sono costituiti da quelle a geometria tetraedrica (numero di coordinazione 4), cui appartiene anche il carbonio sp3, da quelle a geometria piana quadrata (numero di coordinazione 4) e da quelle a geometria ottaedrica (numero di coordinazione 6).
Sostituzioni tetraedriche
Le sostituzioni che avvengono presso un atomo a coordinazione tetraedrica sono molto diffuse e interessano sia elementi dei gruppi principali del sistema periodico (non di transizione) sia i metalli di transizione. Rimanendo nello stesso periodo del carbonio, nei primi due gruppi il litio e il berillio sono caratterizzati da una geometria tetraedrica, mentre il legame assume un carattere elettrovalente; negli elementi dei gruppi successivi (boro, carbonio, azoto, ecc.) il legame ha invece carattere, almeno prevalentemente, covalente. La coordinazione tetraedrica si ritrova anche in molti importanti casi di composti di elementi dei periodi successivi, per esempio alluminio, gallio, silicio, germanio, stagno, fosforo, arsenico e alogeni.
Tra i metalli di transizione si trovano sostituzioni su strutture tetraedriche caratterizzate sia da legami essenzialmente elettrovalenti (TiCl4, MnCl42−, FeC4−, Co Cl42−, NiCl42−), sia da legami covalenti (Ni(CO)4, Pt(PPh3)4, Cu(PPh3)3I) aventi metalli a configurazione d10. A questi esempi si aggiungono importanti e diffusi substrati, quali gli ossianioni MnO4− , CrO42− , Cr2O72−.
Del carbonio tetraedrico si è già trattato, ma si deve osservare che esistono varie differenze di proprietà tra carbonio e silicio, tali da giustificare per questi due elementi un quadro di reattività ben distinto, al di là di analogie formali. Si hanno differenze significative nel più grande raggio covalente del silicio, nelle energie di dissociazione generalmente minori, nell'accessibilità energetica di orbitali d vacanti e, infine, nei numeri di coordinazione. Numeri di coordinazione inferiori a 4 sono estremamente improbabili nel silicio, a differenza del carbonio (come nel caso dei carbocationi), mentre una covalenza superiore a 4 è frequente. Una diretta conseguenza di queste caratteristiche della coordinazione è l'assenza di meccanismi dissociativi nella sostituzione al silicio tetraedrico. Laddove (C6H5)3CCl subisce la solvolisi con un meccanismo dissociativo ‒ promosso dalla stabilità del carbocatione intermedio, (C6H5)3C+ ‒ l'analogo composto del silicio, (C6H5)3SiCl, reagisce con un meccanismo bimolecolare.
D'altra parte, neanche il meccanismo bimolecolare che si ritrova di norma in tutte le sostituzioni nucleofile dei composti del silicio è generalmente analogo a quello indicato dal simbolo SN2 per i composti del carbonio. Vi possono essere diversi modi di reagire, che il substrato sceglie in base a differenze di condizioni, anche modeste.
Un altro centro tetraedrico molto studiato è il fosforo, specie in molecole del tipo R3PO. Anche in questo caso prevalgono meccanismi associativi e percorsi stereochimici che, a seconda dei casi, permettono l'inversione o la conservazione. Tuttavia, anche se meno frequenti e meno ben dimostrati, esistono esempi di processi dissociativi, come negli ossianioni quale Cl2PO2−.
Sostituzioni presso centri a struttura ottaedrica
La struttura ottaedrica è tra le più diffuse e frequenti di tutta la chimica. Essa caratterizza la distribuzione di atomi e gruppi atomici intorno agli elementi dei più diversi settori del sistema periodico, legati con legami chimici sia ionici (come nei cationi Mg(H2O)62+, Fe(H2O)63+), sia covalenti (come nelle molecole SF6 e Cr(CO)6 e nell'anione Fe(CN)64−), ma è particolarmente favorita nella configurazione d6 degli elementi di transizione, quali V(−I), Cr(0), Mo(0), Mn(I), Fe(II), Ru(II), Co(III), Rh(III), Ni(IV), Pd(IV), Pt(IV). In tutte le sostituzioni note dei composti ottaedrici, si nota una notevole uniformità di meccanismi di reazione, dove domina, sia pure con leggere variazioni, il carattere dissociativo, anche se molte configurazioni elettroniche sono insature e potrebbero permettere processi associativi. Generalmente, le reazioni dipendono poco dalla concentrazione e dalla natura del reagente nucleofilo: questa è una prima caratteristica sperimentale a favore del meccanismo dissociativo. Senza entrare nei dettagli della cinetica chimica, le reazioni possono andare da un meccanismo di interscambio a un meccanismo dissociativo. Al primo tipo appartiene la reazione del complesso Co(NH3)5 H2O3+ con nucleofili anionici Y−: si ammette che in essa abbia luogo un equilibrio preliminare (rapido), nel quale il reagente prende posto nella sfera di solvatazione del complesso, cui segue lo scambio di posto tra il legante uscente e il legante entrante che si trova nelle immediate vicinanze:
[22] formula[23]
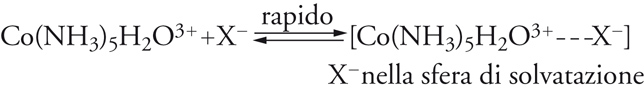
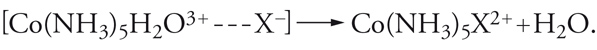
Di tipo dissociativo è la reazione del complesso Co(CN)5H2O2− con analoghi reagenti, dove nel pre-equilibrio avviene una dissociazione completa seguita dall'attacco del reagente sull'intermedio pentacoordinato:
[24] Co(CN)5H2O2− →← Co(CN)52− + H2O
[25] Co(CN)52− + X− → Co(CN)5X3−.
La differenza tra i due modi di reagire è più di misura che di qualità, poiché nel secondo caso l'intermedio pentacoordinato ha un tempo di vita media finito e sufficientemente lungo da potersi equilibrare con la sfera di solvatazione circostante.
Considerazioni e prospettive sullo stato delle conoscenze delle reazioni chimiche
L'approfondimento delle conoscenze sulla natura delle reazioni chimiche ha creato una piattaforma culturale comune a settori tradizionalmente rimasti separati nelle divisioni classiche della chimica (inorganica, organica, ecc.). Sebbene certe caratteristiche di struttura e di reattività siano prevalenti per determinati elementi e per i loro composti, esistono caratteristiche comuni, per qualità se non per misura, da parte dei diversi elementi. In una visione unificata della chimica, ha sempre meno senso tenere distinta la chimica organica dalla chimica inorganica se si considera che il carbonio è in grado di combinarsi con tutti gli altri elementi del sistema periodico producendo una grandissima varietà di composti che non sono né tipicamente organici né tipicamente inorganici. I metodi della chimica fisica servono ugualmente bene tanto al progresso della chimica del carbonio quanto a quello della chimica degli altri elementi.
Ci si può chiedere in quali direzioni si prevede un avanzamento dello studio delle reazioni chimiche negli anni avvenire. Sostanziale è certamente lo sviluppo dei progressi delle tecnologie sperimentali. La comparsa di tecniche che permettono di studiare reazioni estremamente rapide dischiude nuovi campi di studio, specialmente tra le reazioni inorganiche. I risultati così ottenuti possono essere confrontati con quelli derivanti dall'applicazione dei calcoli quantomeccanici che sono ormai in grado di descrivere le caratteristiche degli atti molecolari elementari. Il ruolo delle interazioni solvente-solvente nelle reazioni chimiche è uno dei problemi di fondo della reattività chimica in soluzione. Esso è meglio compreso se confrontato con il comportamento delle reazioni in fase gassosa.
Un'altra direzione è lo studio delle reazioni organometalliche e dei meccanismi di catalisi omogenea che passano attraverso intermedi attivi organometallici. Approfondimenti devono essere condotti anche sulla natura delle reazioni eterogenee che offrono campi di indagine ricchi di conseguenze, dalla catalisi micellare alle reazioni che avvengono sulla superficie dei solidi; sulle reazioni fotocatalizzate e sulle reazioni chimiche che permettono di immagazzinare energia solare nei sistemi materiali. Ma se l'integrazione della chimica è un importante fattore di sviluppo, non meno importante è la ricerca nelle aree di interfaccia della chimica con altre discipline: la fisica, la biologia e l'ingegneria. Occorrerà dare maggior riconoscimento all'importanza del contributo della chimica alla soluzione dei problemi connessi con i sistemi complessi, se si considera che il loro comportamento (il motore di un razzo, la cellula vivente, l'atmosfera inquinata, ecc.) dipende in modo critico dalle proprietà chimiche e, in modo particolare, dalle reazioni chimiche.
Bibliografia
American Chemical Society 1971: Preliminary report: International conference on education in chemistry, "Journal of chemical education", 48, 1971, pp. 3-13.
Basolo, Pearson 1967: Basolo, Fred - Pearson, Ralph G., Mechanisms of inorganic reactions, New York, Wiley, 1967.
Chapman, Shorter 1972: Chapman, Norman B. - Shorter, John, Advances in linear free energy relationships, London-New York, Plenum, 1972.
Coetzee, Ritchie 1969: Coetzee, Johannes F. - Ritchie, Calvin D., Solute-solvent interactions, New York, Dekker, 1969.
Fendler, Fendler 1975: Fendler, Janos H. - Fendler, Eleanor J., Catalysis in micellar and macromolecular systems, New York, Academic Press, 1975.
Frost, Pearson 1961: Frost, Arthur A. - Pearson, Ralph G., Kinetics and mechanism, New York, Wiley, 1961.
Glasstone 1941: Glasstone, Samuel - Laidler, Keith J. - Eyring, Henry, The theory of rate processes, New York, McGraw-Hill, 1941.
Illuminati 1971: Illuminati, Gabriello, Chimica organica, Roma, Libreria eredi V. Veschi, 1971.
Lewis, Randall 1923: Thermodynamics and the free energy of chemical substances, edited by Gilbert N. Lewis, Merle Randall, New York-London, McGraw-Hill, 1923.
March 1977: March, Jerry, Advanced organic chemistry, 2. ed., New York, McGraw-Hill, 1977.
McManus 1973: Organic reactive intermediates, edited by Samuel P. McManus, New York, Academic Press, 1973.
Melander 1960: Melander, Lars, Isotope effects on reaction rates, New York, Ronald, 1960.
Solov'ev 1976: Solov'ev, Jurij I., L'evoluzione del pensiero chimico, Milano, Mondadori, 1976.
Tobe 1972: Tobe, Martin L., Inorganic reaction mechanisms, London, Nelson, 1972.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


