Fluorescenza
Enciclopedia della Scienza e della Tecnica (2008)
Fluorescenza
La microscopia a fluorescenza, come approccio sperimentale per l’osservazione diretta di fenomeni biologici in cellule vive, ha registrato negli ultimi anni un marcato impulso, che ha beneficiato di due fattori: il progresso tecnologico degli strumenti di indagine, con lo sviluppo della microscopia confocale e dell’analisi matematica di immagini di fluorescenza tradizionale (imaging digitale), e lo sviluppo di sonde specifiche per marcare e seguire dinamicamente strutture cellulari o parametri fisiologici.
Due tipi di sonde, per la loro facilità d’uso e per le importanti applicazioni, hanno riscosso notevole successo: gli indicatori fluorescenti dello ione Ca2+, che hanno esteso la determinazione di questo ione a tutti i tipi cellulari in coltura e in situ, e le proteine ricombinanti, che offrono il grande vantaggio di poter essere indirizzate selettivamente a distretti cellulari di interesse, tramite l’aggiunta di segnali di localizzazione specifici alla sequenza della proteina.
Nel corso degli ultimi decenni, un numero crescente di ricercatori ha intrapreso l’osservazione diretta dei processi essenziali della vita di una cellula, quali, per esempio, il movimento, la secrezione, la contrazione, la divisione o la morte. Almeno tre fattori hanno contribuito alla diffusione di questo approccio sperimentale. Il primo, indiretto, è stato l’enorme sviluppo della biologia molecolare, che ha permesso la ‘dissezione’ di processi complessi, come quelli sopra citati, e l’identificazione degli elementi molecolari che vi partecipano. In molti casi, l’analisi molecolare ha rivelato scenari complessi, identificando isoforme diverse della stessa proteina con distribuzione tissutale e proprietà funzionali distinte; tuttavia, il ruolo reale dei diversi attori molecolari in situ è ancora oscuro. Allo stesso tempo, le tecniche di biologia molecolare hanno aperto la possibilità di esprimere per via ricombinante una proteina eterologa, ossia di aggiungere una proteina specifica al repertorio naturale di una cellula. Si può così dotare di una particolare proteina (per es., una molecola segnale come una chinasi) una cellula che ne è sprovvista, oppure, esprimendo una forma mutata di una proteina endogena, verificare l’effetto di specifiche alterazioni biochimiche. Con un approccio un po’ più complesso, infine, si può distruggere il gene di una particolare proteina e studiare l’effetto della sua ablazione. In tutti i casi, lo studio diretto del processo di interesse nella cellula modificata ha una grande importanza e utilità.
Il secondo fattore di rilievo è stato lo sviluppo tecnologico, che ha permesso di realizzare microscopi e sistemi di analisi di immagine sempre più complessi. Risale agli anni Novanta del Novecento l’affermazione della microscopia confocale e lo sviluppo di algoritmi che permettono di calcolare, anche su immagini di fluorescenza tradizionale, il contributo al segnale dovuto alle porzioni non a fuoco del campione. Questi due approcci permettono di ottenere immagini a risoluzione e contrasto elevati anche da campioni complessi, come animali interi.
Un terzo elemento decisivo per sfruttare appieno le nuove tecnologie di analisi di immagine è la disponibilità di sonde che permettono di marcare, all’interno di una cellula viva, elementi subcellulari di interesse (organuli, proteine ecc.) o di misurare parametri cellulari importanti, come la concentrazione dello ione Ca2+, secondo messaggero intracellulare. Anche se la prima dimostrazione del ruolo degli ioni Ca2+ nel controllo di una funzione cellulare (la contrazione del muscolo cardiaco) risale a oltre un secolo fa, l’applicazione di questo meccanismo – che rappresenta oggi una nozione consolidata in biologia – a un’ampia varietà di funzioni e di tipi cellulari ha dovuto attendere lo sviluppo di specifici sitemi di misura della concentrazione di calcio ionizzato all’interno di una cellula. A questo scopo, sono risultate di grande utilità due categorie di molecole fluorescenti che hanno rivoluzionato il campo di studio della microscopia a fluorescenza: gli indicatori fluorescenti di parametri cellulari, molecole non proteiche intrappolabili nel citoplasma, di cui gli indicatori di Ca2+ sono l’esempio più importante, e la green fluorescent protein (GFP, proteina a fluorescenza verde), una proteina della medusa Aequorea victoria. Con esse, va ricordata la fotoproteina equorina, che, pur essendo una proteina chemiluminescente e non fluorescente, ha un rilevante significato storico, in quanto è stata la prima proteina ricombinante utilizzata per la misura di un parametro fisiologico importante (la concentrazione di Ca2+) e ha messo in luce le straordinarie potenzialità di questo approccio sperimentale.
Gli indicatori fluorescentidello ione Ca2+
Negli ultimi decenni del XX sec. si sono registrati due grandi avanzamenti metodologici nel campo dei sistemi di misurazione della concentrazione dello ione calcio all’interno delle cellule. Il primo è stato l’isolamento di molecole naturali, come la fotoproteina equorina, o di sintesi, come gli indicatori metallocromici arsenazo III o antipirilazo III, in grado di misurare in maniera attendibile la concentrazione di Ca2+. Questi indicatori hanno avuto un ruolo importante nello studio del ruolo dello ione Ca2+ come secondo messaggero intracellulare. Per esempio, uno dei fenomeni tuttora più studiati della segnalazione via Ca2+, le oscillazioni ritmiche della concentrazione di Ca2+ indotte da una varietà di stimoli, è stato identificato e caratterizzato grazie a studi eseguiti con equorina. Tuttavia, questi studi risentivano di una limitazione sperimentale importante: l’indicatore di Ca2+ doveva essere introdotto direttamente all’interno della cellula per microiniezione, perché né una proteina né una molecola carica, come un indicatore metallocromico, attraversano la membrana di una cellula viva. Di necessità, quindi, questo approccio sperimentale era limitato a un numero alquanto ridotto di tipi cellulari, nei quali era praticabile la microiniezione dell’indicatore.
La situazione è radicalmente cambiata con lo sviluppo da parte di Roger Y. Tsien e dei suoi collaboratori di un secondo metodo di misura della concentrazione dello ione calcio intracellulare: quello degli indicatori fluorescenti intrappolabili nel citoplasma. Questi indicatori, oltre a rivoluzionare lo studio dell’omeostasi intracellulare di Ca2+, sono stati il prototipo di una varietà di indicatori per altri parametri fisiologici, quali il pH, la concentrazione di Na+ o di Mg2+ e così via.
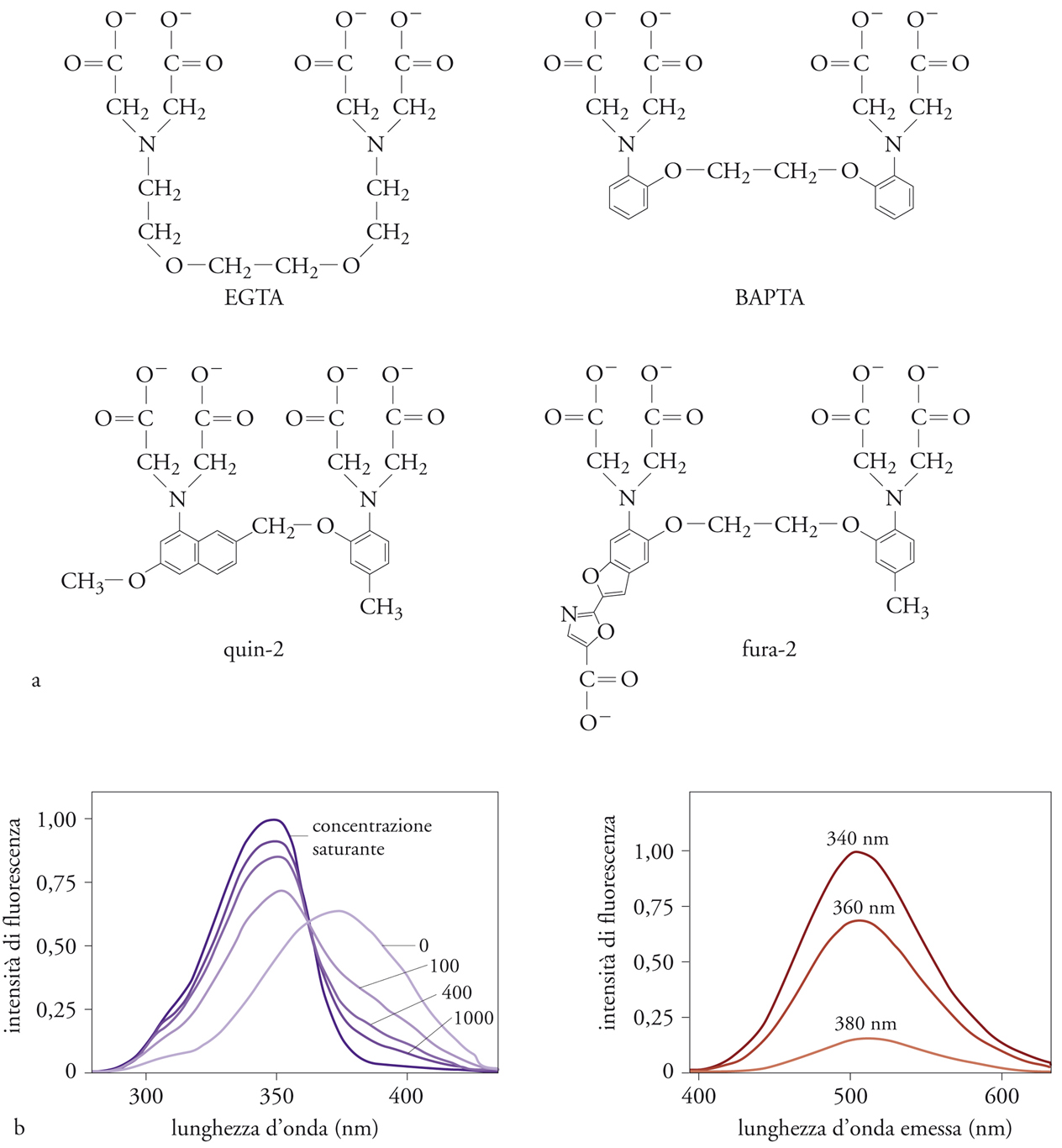
Gli elementi essenziali di un indicatore sono la selettività, la qualità del segnale e la facilità d’uso. Gli indicatori fluorescenti di Ca2+, il cui precursore è stata la molecola quin-2, ma che attualmente annoverano decine di composti diversi, hanno come punto di partenza un chelante selettivo per Ca2+, come l’EGTA o il BAPTA (fig.1). La porzione della molecola che lega Ca2+ è costituita da una gabbia carbossilica che si adatta esattamente alle dimensioni dello ione: da qui ha origine la selettività. Alla gabbia carbossilica è associato un gruppo fluoroforo, che conferisce alla molecola una fluorescenza dipendente dal legame di Ca2+ con la gabbia carbossilica.
Per la loro struttura, e in particolare per la presenza dei gruppi carbossilato che legano lo ione Ca2+, anche questi indicatori non attraversano liberamente le membrane cellulari. Per risolvere questo problema, la molecola attiva è stata ulteriormente modificata esterificando (e quindi neutralizzando) i gruppi carichi. In questa forma la molecola, incapace di legare Ca2+, attraversa la membrana plasmatica delle cellule e raggiunge il citoplasma, dove enzimi che fanno parte del corredo normale della cellula (le esterasi) idrolizzano l’estere e ripristinano la forma attiva della molecola. Come si può comprendere, questo semplice accorgimento è la chiave del successo di tali indicatori. Infatti, il loro uso è molto semplice: basta incubare le cellule di interesse con la forma esterificata dell’indicatore per un tempo relativamente breve (di solito 20÷30 minuti) per ‘caricarne’ all’interno della cellula una quantità tale da permettere la determinazione di Ca2+. Si rimuove quindi l’indicatore dal terreno di coltura, eliminando così una sorgente di fluorescenza spuria, e per le misure si utilizza la frazione di indicatore attivo intrappolata, grazie all’azione delle esterasi, nel citoplasma delle cellule.
Gli indicatori fluorescenti di nuova generazione
Rispetto al primo indicatore fluorescente prodotto da Tsien, gli indicatori sviluppati negli anni successivi sono dotati di diverse caratteristiche chimiche, che li rendono più adatti agli studi fisiologici.
Anzitutto, la maggiore intensità molare di fluorescenza permette di utilizzare quantità minori di indicatore, riducendone così il ruolo di tampone delle concentrazioni di Ca2+ citosoliche. Infatti, più intensa è la fluorescenza intrinseca, minore è la quantità di indicatore necessaria alla misura, e quindi minore è il turbamento dell’omeostasi intracellulare dello ione. In media, per una misura efficace della concentrazione di Ca2+ con quin-2, è necessario ‘caricare’ le cellule con una concentrazione di indicatore pari a circa 0,5÷1,0 mM, mentre per il fura-2, l’indicatore oggi più utilizzato, sono sufficienti concentrazioni 10 volte inferiori. In secondo luogo, la minore affinità per Ca2+ rende i nuovi indicatori più adatti a misurare concentrazioni di 1÷2 μM (valori di concentrazione frequentemente raggiunti nel citoplasma di cellule stimolate), alle quali invece la fluorescenza del quin-2 è saturata. Inoltre, nel caso dei nuovi indicatori, il legame dello ione Ca2+ non causa solo una variazione quantitativa della fluorescenza emessa, ma anche una variazione spettrale (a seconda dell’indicatore, nello spettro di eccitazione o di emissione). In altre parole, sia la forma libera dell’indicatore che la forma complessata contenente lo ione Ca2+ sono intensamente fluorescenti, con caratteristiche diverse. Per esempio, all’aumentare della concentrazione di Ca2+ cui è esposto, l’emissione di luce (a 505 nm) del fura-2 aumenta se illuminato a 340 nm, mentre diminuisce se illuminato a 380 nm (fig. 1B). La concentrazione di Ca2+ può quindi essere ricavata dal rapporto dell’intensità di fluorescenza ottenuta illuminando il campione alle due lunghezze d’onda, rendendo così la misura indipendente dalla quantità assoluta di indicatore disponibile, che invece influenza marcatamente i risultati ottenuti con misure a una singola lunghezza d’onda. Questa proprietà, che ha rappresentato un avanzamento metodologico significativo, è fondamentale in esperimenti di imaging a singola cellula, date le disomogeneità di caricamento nelle varie porzioni delle cellule dovute al differente spessore, al diverso contenuto di altre strutture e ad altri parametri.
Il cambiamento Ca2+-dipendente dello spettro di emissione, caratteristico di alcuni nuovi indicatori (per es., indo-1), risulta particolarmente utile per l’uso del microscopio confocale. In questo strumento, infatti, l’eccitazione del campione avviene mediante un laser, e perciò risulta relativamente complesso variare la lunghezza d’onda di eccitazione e quindi utilizzare indicatori con variazioni Ca2+-dipendenti nello spettro di eccitazione, come il fura-2. Viceversa, con indicatori come indo-1, utilizzando due diversi fotomoltiplicatori che raccolgono, grazie a opportuni sistemi di filtri dicroici, la luce a diversa lunghezza d’onda, è possibile svolgere lo stesso tipo di misura (rapporto tra lunghezze d’onda Ca2+-sensibile e Ca2+-non sensibile) con il microscopio confocale. Un ulteriore vantaggio offerto da alcuni nuovi indicatori, quale per esempio il fluo-3, sta nel fatto che essi vengono eccitati con luce visibile. Viceversa, il quin-2 e il fura-2 utilizzano, come sorgente di eccitazione, la luce UV, la quale, però, modificando macromolecole biologiche, può danneggiare le cellule o causare effetti collaterali indesiderati. A tali lunghezze d’onda, inoltre, l’autofluorescenza cellulare (dovuta a composti endogeni come NADH) risulta assai marcata.
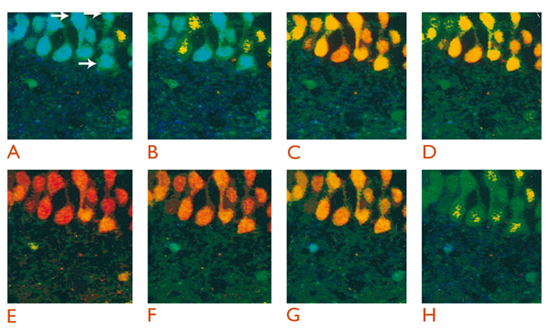
L’esperimento illustrato in fig. 2 rappresenta un esempio dell’applicazione degli indicatori fluorescenti alla misura della concentrazione di Ca2+. Esso si riferisce infatti a misure della variazione della concentrazione di Ca2+ citoplasmatico in astrociti e neuroni in situ, cioè in cellule appartenenti a sezioni pressoché integre di tessuto cerebrale (nel caso specifico la regione ippocampale CA1). Nell’esperimento mostrato, sezioni sottili, di spessore pari a circa 200 μm, sono state ottenute dal cervello di ratti neonati attraverso l’uso di un vibratomo e caricate con l’indicatore fluorescente indo-1, con una procedura simile a quella comunemente impiegata per le cellule in coltura. Al termine dell’incubazione, la sezione cerebrale è stata trasferita nel campo di indagine di un microscopio confocale, che, in un campione complesso come una porzione di tessuto, permette di analizzare selettivamente il segnale di fluorescenza proveniente da singole cellule. I cambiamenti della concentrazione citoplasmatica di Ca2+, che nel caso dell’indo-1 si traducono in cambiamenti del rapporto tra l’intensità di fluorescenza emessa a due lunghezze d’onda distinte (405 e 485 nm), sono espressi in una scala di pseudocolori. In presenza di uno stimolo depolarizzante (KCl a concentrazione 40 mM), le cellule piramidali mostrano un evidente aumento della concentrazione di Ca2+ dovuto all’entrata di tale ione attraverso i canali per il calcio dipendenti da voltaggio. Con la sospensione dello stimolo, le cellule riportano la concentrazione di Ca2+ citoplasmatica a livelli basali.
Le sonde proteiche: l’equorina, una sonda di Ca2+ a destino intracellulare specifico
L’ampia diffusione delle tecniche di biologia molecolare, con la possibilità di modificare l’espressione genica delle cellule di interesse, eventualmente inducendo la produzione di proteine esogene, è stata il motore dell’impressionante espansione nell’uso di sonde proteiche in biologia cellulare. Due tipi di sonde proteiche attualmente utilizzate derivano dall’ampia varietà di organismi bioluminescenti presenti in natura. La prima categoria è quella delle proteine luminescenti, ossia proteine che emettono luce, spesso in risposta a variazioni di parametri di interesse fisiologico, quali le concentrazioni di ATP o di Ca2+. Poiché le cellule di mammifero non possiedono molecole luminescenti, l’uso delle proteine luminescenti in studi di biologia cellulare è di solito associato a un eccellente rapporto segnale/rumore. Viceversa, poiché il segnale rispecchia unicamente l’emissione di luce da parte della proteina ricombinante, esso è piuttosto debole e quindi può essere raccolto o integrando il segnale proveniente da almeno 103÷104 cellule o, a livello di singola cellula, con strumenti di analisi di immagine a elevatissima sensibilità e costo. Il secondo gruppo di sonde proteiche, che incontra una utilizzazione sempre più ampia, è dato dalle proteine fluorescenti. Tra queste, protagonista indiscussa è la green fluorescent protein di Aequorea victoria, che sarà discussa estesamente in seguito.
Come accennato precedentemente, l’equorina, così come gli indicatori metallocromici, è stata gradualmente sostituita, nello studio dell’omeostasi dello ione Ca2+, dai coloranti fluorescenti sviluppati da Tsien e dai suoi collaboratori. I nuovi coloranti, infatti, oltre che di uso più semplice, possono essere impiegati su un’ampia varietà di tipi cellulari e permettono, grazie all’intenso segnale di fluorescenza, di utilizzare microscopi a fluorescenza tradizionali o confocali per ottenere immagini a elevata risoluzione delle variazioni della concentrazione di Ca2+ indotte da vari stimoli fisiologici, rivelando così la complessità spazio-temporale di questa modalità di segnalazione.
L’isolamento del cDNA dell’equorina, tuttavia, ha di nuovo valorizzato questa proteina come strumento per misurare la concentrazione dello ione Ca2+ in cellule vive. Innanzitutto, la disponibilità del cDNA risolve il problema dell’introduzione di questa proteina nelle cellule. Transfettare le cellule, ossia introdurre un plasmide che le induca a produrre una proteina estranea, quale l’equorina, è una procedura semplice ed efficace. È possibile introdurre per transfezione l’equorina in cellule assai diverse per struttura e origine embriologica, come le linee stabilizzate epiteliali (Re La, CRO), le colture primarie di muscolo scheletrico e di tessuto nervoso, e così via. Semplificata così l’utilizzazione, la rivalutazione dell’equorina è dipesa non solo da alcune caratteristiche funzionali favorevoli, quali l’eccellente rapporto segnale/rumore, l’ampio spettro di concentrazioni di Ca2+ misurabili, l’efficacia a concentrazioni assai ridotte, che riduce l’effetto tampone sulla concentrazione di Ca2+, ma, soprattutto, dal grande vantaggio offerto da una proteina, ossia la possibilità di controllarne in maniera selettiva la localizzazione. È noto infatti che la destinazione delle proteine endogene di una cellula dipende, nella maggior parte dei casi, dalla presenza, nella sequenza amminoacidica della proteina stessa, di sequenze segnale riconosciute da specifici sistemi di direzionamento e che queste sequenze sono necessarie e sufficienti a determinare la corretta localizzazione. Quando, con tecniche di biologia molecolare, si aggiungono queste sequenze a proteine estranee, esse acquisiscono la distribuzione cellulare determinata dalla sequenza segnale. Nel caso specifico, fondendo cDNA di proteine diverse è stato quindi possibile costruire proteine chimeriche costituite dalla fotoproteina equorina e da porzioni di proteine di mammifero che contengono specifiche sequenze di indirizzamento. Ciascuna proteina chimerica, espressa per via ricombinante in colture cellulari, mantiene la capacità dell’equorina di misurare Ca2+, ma si localizza esclusivamente nel compartimento cellulare deciso dalla sequenza segnale. Questo approccio ha permesso di costruire sonde per Ca2+ che, a differenza dei coloranti fluorescenti, si localizzano selettivamente in un particolare distretto cellulare.
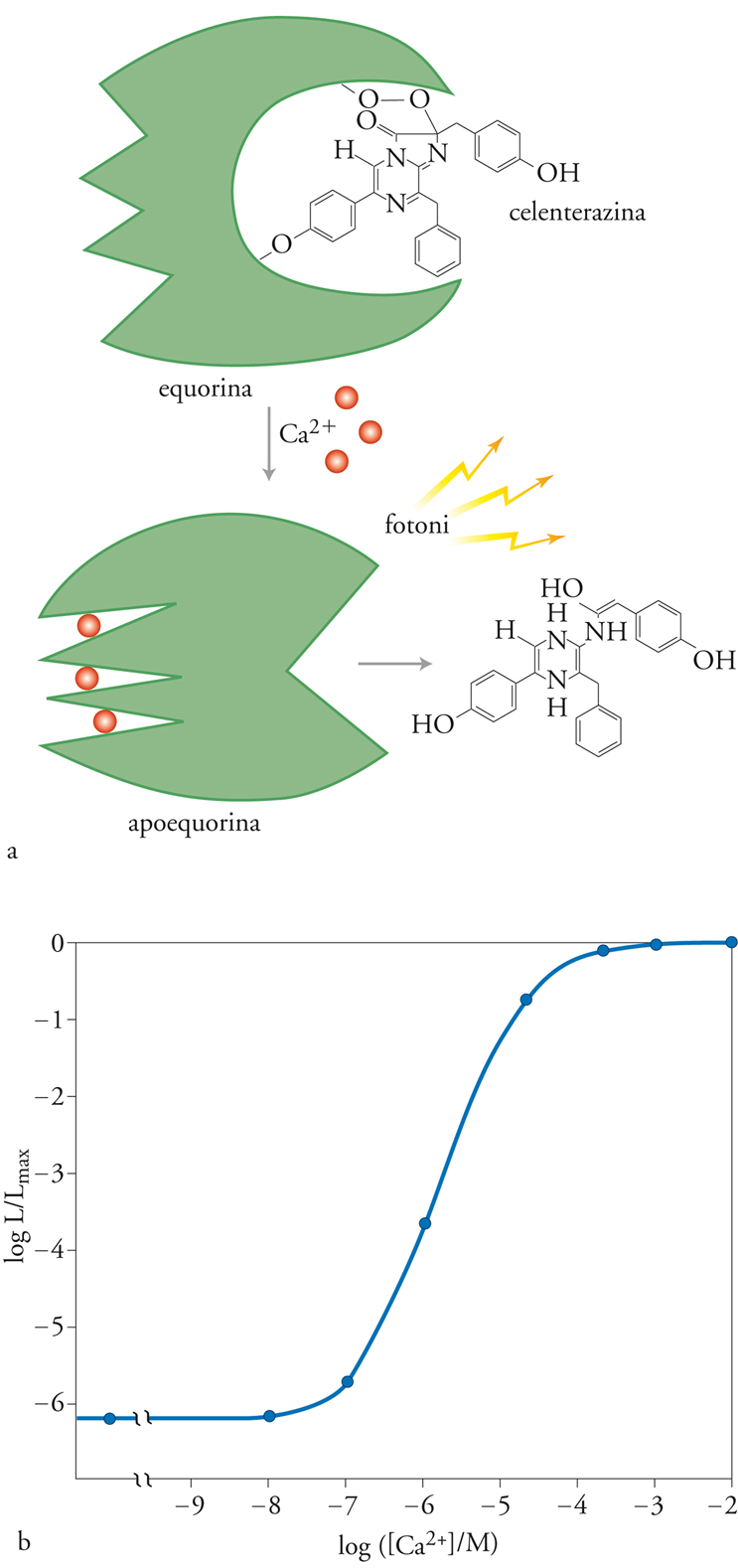
In fig. 3 è mostrato, in alto, lo schema della reazione di emissione di luce da parte dell’equorina. L’equorina è una proteina di circa 21 kDa di diverse specie di Aequorea che, in forma attiva, ha legato un gruppo prostetico idrofobico, la celenterazina. Quando ioni Ca2+ si legano a tre siti di legame ad alta affinità (omologhi a quelli presenti in altre proteine che legano Ca2+, come la calmodulina), l’equorina va incontro a una reazione irreversibile, in cui viene modificato il gruppo prostetico ed emesso un fotone. La velocità della reazione dipende dalla concentrazione di Ca2+ cui l’equorina è esposta, come dimostra il grafico della parte inferiore della fig. 3, relativo alla relazione che intercorre tra la concentrazione di Ca2+ e la frazione di consumo dell’equorina, espressa dal rapporto tra l’intensità della luce emessa a una data concentrazione e il valore massimo di emissione corrispondente a concentrazioni saturanti di Ca2+. In particolare, nell’intervallo fisiologico di valori della concentrazione di Ca2+ (10−7÷10−5 M), l’emissione di luce è proporzionale alla seconda o terza potenza della concentrazione di Ca2+.
Determinazione analitica di Ca2+ in vari scomparti cellulari
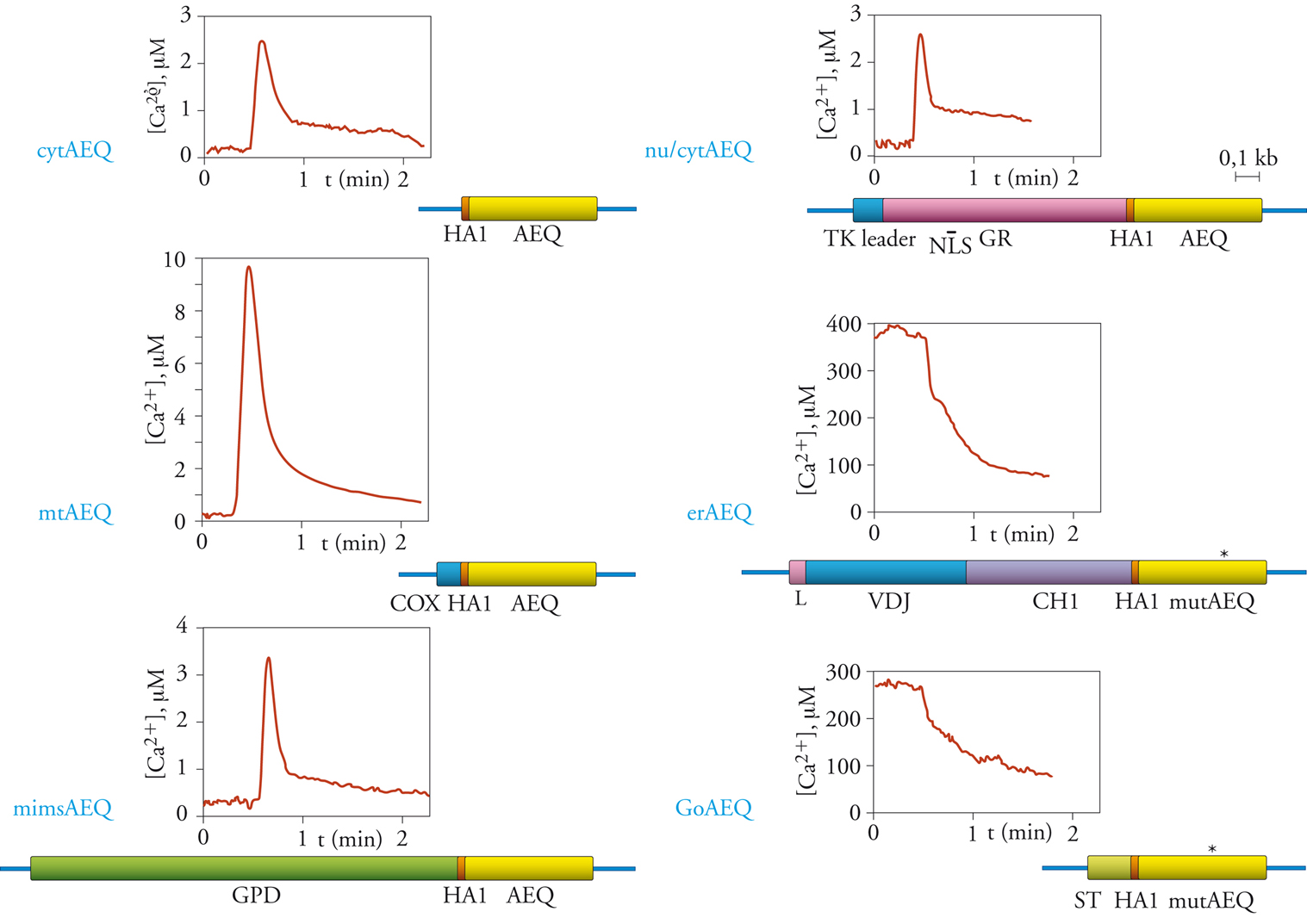
La fig. 4 presenta i cDNA chimerici costruiti per direzionare l’equorina a diversi compartimenti intracellulari e le determinazioni di Ca2+ eseguite in cellule HeLa con questi costrutti.
Citoplasma. - Il costrutto impiegato per misurare la concentrazione di Ca2+ in questo compartimento (cytAEQ) corrisponde sostanzialmente al cDNA nativo dell’equorina, con l’unica aggiunta della breve sequenza amminoacidica HA1, derivata dalla proteina emoagglutinina del capside del virus dell’influenza, riconosciuta da un anticorpo monoclonale. Questa modifica, ossia l’aggiunta di un epitopo forte a una proteina con deboli proprietà immunogeniche (epitope tagging, marcatura con un epitopo), è utile per riconoscere la proteina ricombinante in cellule transfettate. Nella traccia sperimentale corrispondente (fig. 4), le cellule transfettate sono state trattate con una sostanza (istamina) in grado di stimolare le cellule inducendo un aumento della concentrazione citoplasmatica dello ione Ca2+. Ciò avviene per azione su recettori accoppiati, attraverso una proteina G specifica, a un enzima effettore (la fosfolipasi c), che produce il messaggero intracellulare inositolo-1,4,5-trisfosfato (IP3), il quale, a sua volta, agendo su canali situati sulla membrana del reticolo endoplasmatico, causa il rilascio di Ca2+ da questo compartimento al citoplasma. In accordo con risultati ottenuti con gli indicatori fluorescenti, dalla traccia presentata si vede che la concentrazione citoplasmatica di Ca2+, che a riposo è circa 0,1÷0,2 μM, aumenta, dopo l’aggiunta di istamina, fino a valori di circa 2,5 μM, e poi scende, stabilizzandosi a un valore più elevato di quello di partenza, che viene mantenuto per il tempo della stimolazione cellulare.
Mitocondri. - Quasi tutte le proteine mitocondriali, a eccezione dei tredici polipeptidi codificati dal genoma mitocondriale, sono codificate dal nucleo, tradotte su ribosomi citoplasmatici e successivamente importate nei mitocondri. Nella maggior parte dei casi, il riconoscimento come proteina mitocondriale, e quindi l’importazione nell’organello, dipende da una sequenza segnale ricca in amminoacidi basici posta all’estremità amminoterminale del polipeptide che, dopo l’importazione, viene rimossa da proteasi della matrice mitocondriale. Per costruire l’equorina a destino mitocondriale, il cDNA dell’equorina/HA1 viene fuso con la porzione delcDNA di una proteina mitocondriale, la subunità VIII dell’enzima citocromo c-ossidasi, che codifica la sequenza segnale e i primi sei amminoacidi della proteina matura. La proteina chimerica così costruita, l’equorina mitocondriale o mtAEQ, si localizza selettivamente nei mitocondri. Le misure in cellule HeLa, con un protocollo identico a quello riportato sopra, mostrano un risultato assai sorprendente: in seguito a stimolazione con agonisti fisiologici come l’istamina, i mitocondri subiscono variazioni della concentrazione di Ca2+ addirittura superiori a quelle del citoplasma, fino a un valore di picco di circa 10 μM. Il risultato è sorprendente perché i sistemi di trasporto mitocondriali di Ca2+ sono caratterizzati da un’affinità molto bassa, e quindi alle concentrazioni di Ca2+ citoplasmatiche (sia a riposo che dopo stimolazione) ci si aspetterebbe un accumulo mitocondriale di Ca2+ trascurabile. Come sarà mostrato più oltre, l’enigma è stato risolto grazie alla GFP, la proteina fluorescente che, opportunamente direzionata ai mitocondri e al reticolo endoplasmatico, ha dimostrato una relazione strutturale stretta tra l’organulo che funge da deposito intracellulare di Ca2+ e i mitocondri, che quindi, in seguito a stimolazione, ‘sentono’ concentrazioni elevatissime di Ca2+.
Spazio intermembrana dei mitocondri. - I mitocondri sono organuli rivestiti da due membrane, di cui quella esterna liberamente permeabile a ioni e a piccoli soluti e quella interna impermeabile. Per misurare la concentrazione di Ca2+ nello spazio tra le due membrane mitocondriali, che si attendeva diversa da quella della matrice mitocondriale e simile, se non identica, a quella del citoplasma, l’equorina è stata fusa con una proteina residente in questo spazio. La proteina prescelta è stata la glicerofosfatodeidrogenasi (GPD) che, inserita nella membrana interna mitocondriale, espone una voluminosa porzione carbossiterminale nello spazio intermembrana. Tramite fusione dei rispettivi cDNA, l’equorina/HA1 è stata aggiunta all’estremità carbossiterminale della GPD (mimsAEQ). Le misure della concentrazione di Ca2+ in questo distretto in cellule HeLa hanno mostrato che, in seguito a stimolazione con istamina, lo spazio intermembrana dei mitocondri va incontro a un aumento della concentrazione di Ca2+ superiore a quello del citoplasma (valore di picco 3,5 μM contro 2,5 μM del citoplasma). Questo risultato, che potrebbe risultare sorprendente considerato che lo ione Ca2+ viene rilasciato dal reticolo endoplasmatico nel citoplasma e diffonde poi nello spazio intermembrana dei mitocondri, suggerisce che la superficie dei mitocondri sia in realtà esposta non alle concentrazioni medie di Ca2+ del citoplasma, ma a microambienti in cui sono raggiunte concentrazioni di Ca2+ molto elevate. Quest’ultima osservazione è in accordo con i dati ottenuti con le GFP chimeriche (vedi oltre), che hanno mostrato un contatto stretto tra i mitocondri e il reticolo endoplasmatico, il deposito intracellulare di Ca2+ rilasciabile da agonisti.
Nucleo. - La destinazione nucleare delle proteine dipende, anche in questo caso, da una breve sequenza di localizzazione dalle caratteristiche determinate, la cosiddetta sequenza di consenso di localizzazione nucleare (NLS, Nuclear localization sequence) che, a differenza della sequenza mitocondriale, non ha una posizione preferenziale nella proteina e non viene rimossa dopo che questa ha raggiunto la corretta destinazione intracellulare. In alcuni casi, le proteine possono raggiungere il nucleo solo in alcune fasi della vita della cellula, o in alcune condizioni fisiologiche. Questo accade perché la proteina assume in queste condizioni una conformazione particolare ed espone alla superficie la sequenza NLS, che viene così riconosciuta e permette il trasporto nel nucleo. Per esempio, la sequenza NLS del recettore degli ormoni glucocorticoidi (GR) è esposta (e quindi è attiva) solo quando il recettore lega l’ormone; solo in questo caso la proteina trasloca nel nucleo dove esercita la propria azione biologica. L’equorina nucleare (nu/cytAEQ), costruita mediante fusione dei rispettivi cDNA, comprende l’equorina/HA1 e un’ampia porzione del GR, e mantiene la distribuzione intracellulare di quest’ultimo. Essa quindi trasloca nel nucleo solo quando le cellule vengono trattate con ormoni glucocorticoidi, altrimenti resta nel citoplasma delle cellule. La localizzazione intracellulare di questa sonda per Ca2+ dipende perciò dalle condizioni sperimentali. Ciò è risultato molto utile perché ha permesso di adoperare la medesima sonda, evitando così artefatti dovuti a piccole differenze nell’affinità per Ca2+ delle diverse chimere di equorina, per misurare la concentrazione di Ca2+ in due distretti cellulari (citoplasma e nucleo) in cui era lecito attendersi concentrazioni simili di Ca2+, a riposo e sotto stimolazione. I risultati ottenuti mostrano che, in effetti, il nucleo va incontro alle stesse variazioni di Ca2+ indotte da agonista osservate nel citoplasma (il valore di picco raggiunto dopo l’aggiunta di istamina è ca. 2,5 μM), suggerendo che i pori nucleari in vivo siano aperti e che quindi il segnale Ca2+ indotto nel citoplasma si trasmetta al nucleo senza significativi rallentamenti o riduzioni di ampiezza.
Reticolo endoplasmatico. - Le proteine del reticolo endoplasmatico sono trattenute in questo distretto perché possiedono, di solito, un doppio segnale. Una sequenza idrofobica, posta all’estremità amminoterminale, ne causa la traduzione su ribosomi adesi al reticolo endoplasmatico e l’introduzione nel lume del reticolo endoplasmatico stesso. Questo segnale, e questo destino, sono condivisi anche dalle proteine residenti delle altre porzioni del sistema vescicolare intracellulare (apparato del Golgi, vescicole secretorie) e dalle proteine secrete. La ritenzione delle proteine del reticolo endoplasmatico nel compartimento corretto dipende da un secondo segnale, che riporta al reticolo endoplasmatico la quota di proteina che sfugge verso porzioni più distali. Il più noto tra questi segnali di ritenzione è la sequenza KDEL, posta all’estremità carbossiterminale della proteina. Nel caso dell’equorina, questo segnale non poteva essere utilizzato perché la modifica dell’estremità carbossiterminale della fotoproteina ne compromette irreversibilmente le proprietà funzionali. È stata quindi seguita una strategia alternativa, utilizzando un segnale di ritenzione localizzato in una porzione diversa della proteina. Uno di questi è il segnale che permette alle catene pesanti delle immunoglobuline di essere trattenute all’interno del reticolo endoplasmatico fino all’assemblaggio con le catene leggere, progredendo quindi verso la secrezione solo come immunoglobuline mature. Come evidente dalla mappa del costrutto (fig. 4), l’equorina del reticolo endoplasmatico (erAEQ) è derivata dalla fusione dei cDNA della catena pesante delle immunoglobuline con quello della AEQ/HA1. La proteina chimerica comprende, dall’estremità amminoterminale a quella carbossiterminale, la sequenza idrofobica che permette l’introduzione nel reticolo endoplasmatico (sequenza leader o L), le porzioni VDJ e CH1 (quest’ultima comprende la sequenza di ritenzione nel reticolo endoplasmatico) dell’immunoglobulina, l’epitopo HA1 e l’equorina. Per misurare la concentrazione di Ca2+ nel reticolo endoplasmatico, tuttavia, occorre risolvere un secondo problema. Infatti, pur avendo l’equorina uno spettro dinamico ben superiore a quello dei coloranti fluorescenti, esso non permette di misurare accuratamente le elevate concentrazioni di Ca2+ del reticolo endoplasmatico. L’affinità dell’equorina è stata quindi ridotta con tre approcci diversi: la mutazione di uno dei siti di legame di Ca2+, l’uso di un gruppo prostetico modificato, la celenterazina n, e la sostituzione di Ca2+ all’interno delle cellule con lo ione Sr2+, un catione che simula i comportamenti di Ca2+ ma per il quale l’equorina ha un’affinità circa 50 volte inferiore. I risultati ottenuti con equorina mutata e celenterazina n in cellule HeLa stimolate con istamina, come descritto sopra, mostrano un quadro radicalmente diverso da quello dei distretti analizzati finora: nel lume del reticolo endoplasmatico la concentrazione di Ca2+ a riposo è pari a circa 400 μM (ossia più di 1000 volte superiore a quella del citoplasma) e, in seguito a stimolazione con istamina, si riduce rapidamente, per il rilascio attraverso i canali attivati dall’IP3. Questi risultati dimostrano che il reticolo endoplasmatico coincide con i depositi intracellulari di Ca2+ rilasciabile in seguito a stimolazione cellulare.
Apparato del Golgi. - Le sequenze segnale responsabili della localizzazione delle proteine residenti nell’apparato del Golgi sono meno conosciute di quelle dei distretti descritti sopra. È noto tuttavia che una porzione dell’enzima sialiltransferasi, comprendente il segmento transmembrana, contiene l’informazione per il corretto direzionamento all’apparato del Golgi e, se fusa con una proteina eterologa, ne causa la ritenzione in questo organulo cellulare. Per destinare l’equorina all’apparato del Golgi tramite fusione dei relativi cDNA, la fotoproteina è stata aggiunta alla metà amminoterminale della sialiltransferasi, che comprende il segmento transmembrana. Le misure della concentrazione di Ca2+ nel lume dell’apparato del Golgi eseguite con questa chimera di equorina (GoAEQ) hanno fornito risultati interessanti e inaspettati. Come si vede dalla fig. 4, infatti, che mostra un esperimento eseguito su cellule HeLa con il protocollo sperimentale già descritto, la concentrazione di Ca2+ nell’apparato del Golgi a riposo è molto elevata (quasi 300 μM), confrontabile, anche se inferiore, con quella del reticolo endoplasmatico. Inoltre, in seguito a stimolazione con istamina, si ha una rapida riduzione di questo valore, a indicare che l’apparato del Golgi condivide con il reticolo endoplasmatico il ruolo di deposito intracellulare di Ca2+ rilasciabile da agonista, un fenomeno non noto e largamente inatteso.
Le sonde proteiche: la green fluorescent protein
La green fluorescent protein di Aequorea victoria è diventata a partire dagli anni Novanta uno strumento di larghissimo uso in biologia cellulare. Benché fosse noto da almeno due decenni che le proprietà luminose di alcune specie marine dipendono dalla presenza non solo di fotoproteine, che emettono luce attraverso la conversione enzimatica di un cofattore (come l’equorina), ma anche di proteine fluorescenti, che modificano il colore della luce emessa, l’idea della loro utilizzazione è molto recente. Dopo il clonaggio del cDNA della GFP nel 1992, in uno studio illuminante Martin Chalfie e collaboratori hanno mostrato che, grazie alla fluorescenza di GFP, era possibile identificare le cellule neuronali in animali vivi, nel cui genoma era stata inserita una copia del cDNA della GFP sotto il controllo di un promotore genico attivo solo nei neuroni. Questo elegante esperimento non solo ha dimostrato che la semplice espressione ricombinante della GFP produce una proteina intensamente fluorescente, ma ha anche fornito un primo esempio chiaro di applicazione biologica della GFP. La diffusione di questo approccio sperimentale è stata impressionante. Centinaia di laboratori nel mondo utilizzano questa proteina per le più svariate applicazioni di ricerca.
Le ragioni del successo della GFP sono evidenti. Innanzitutto, come discusso sopra, la fluorescenza di GFP non è specie-specifica e non richiede l’aggiunta e la diffusione di cofattori all’interno della cellula transfettata con il cDNA della GFP. La sua semplice espressione ricombinante fornisce quindi un intenso segnale di fluorescenza in batteri, funghi, lieviti, cellule vegetali e animali. Inoltre, le dimensioni limitate della proteina (ca. 27 kDa) ne permettono la fusione con altre proteine di interesse, senza interferire significativamente con l’assemblaggio o la funzione di queste ultime. Infine, l’isolamento di mutanti con caratteristiche spettrali diverse, come sarà discusso tra poco, amplia sensibilmente la gamma delle applicazioni.
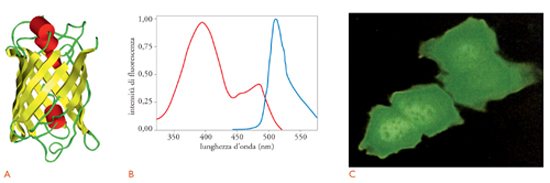
La fig. 5 mostra la struttura tridimensionale della GFP. Come si vede, la proteina è formata da una struttura cilindrica compatta composta da undici foglietti β, che circondano una α-elica che contiene il fluoroforo, costituito dalla ciclizzazione di tre amminoacidi della proteina, Ser(65)-Tyr-Gly. La ciclizzazione è un processo lento che avviene, in presenza di ossigeno, dopo la sintesi della proteina, ed è responsabile della latenza tra la biosintesi della proteina e la comparsa del segnale fluorescente, un ostacolo importante all’utilizzazione della GFP come rivelatore dell’espressione genica.
Il fluoroforo della GFP esiste in due forme chimiche diverse, che sono responsabili dei due picchi di eccitazione della proteina fluorescente (fig. 5B). Il picco a 470 nm (luce blu) è dovuto alla forma anionica del fluoroforo, mentre il picco a 395 nm (luce UV) è dovuto alla forma neutra. Entrambe le forme riemettono poi luce verde (picco a 510 nm). Nella GFP nativa, le due forme del fluoroforo sono in equilibrio, in rapporto 6:1 (forma neutra/forma ionizzata) e quindi il picco di eccitazione è maggiore per la luce UV. Tuttavia, l’illuminazione con luce UV determina, grazie a un processo noto con il nome di fotoisomerizzazione, la ionizzazione del fluoroforo e quindi la riduzione del segnale fluorescente, mentre aumenta quello rilevabile per illuminazione con luce blu.
La fig. 5C mostra un’immagine di fluorescenza di cellule HeLa, che esprimono il cDNA nativo della GFP, illuminate con luce UV. Si può osservare una fluorescenza verde intensa, distribuita all’intero citoplasma della cellula ed estesa anche al nucleo, e ciò conferma che la GFP, proteina eterologa, non possiede segnali di localizzazione specifici. Anche la diffusione al nucleo non è sorprendente, date le piccole dimensioni della GFP, inferiori al limite di esclusione nucleare (ca. 60 kDa).
I mutanti della GFP
Sin dalla prima descrizione dell’uso della GFP ricombinante e delle sue potenzialità, è iniziato l’isolamento di mutanti, con proprietà di fluorescenza migliori o comunque diverse rispetto alla proteina nativa. La risoluzione della struttura tridimensionale, più recente, ha ulteriormente favorito e accelerato la possibilità di isolare mutanti utili. Senza entrare in dettaglio, discuteremo brevemente le classi principali di mutanti di GFP, con particolare enfasi su quelli utili negli studi di biologia cellulare.
Le GFP delete. - In questa direzione, i risultati ottenuti sono stati largamente insoddisfacenti. Era infatti auspicabile poter diminuire le dimensioni della GFP, per diminuirne l’ingombro sterico e quindi il possibile ostacolo funzionale quando essa viene fusa con proteine di interesse allo scopo di visualizzarle. La delezione progressiva di amminoacidi a partire dalle due estremità ha invece mostrato che possono essere eliminati solamente sette amminoacidi all’estremità carbossiterminale e addirittura un solo amminoacido a quella amminoterminale senza interferire drasticamente con le proprietà di fluorescenza. In altre parole, solo la proteina intera è funzionale, come forse si poteva predire dalla sua complessa struttura tridimensionale.
I mutanti brillanti. - Incrementare il segnale della GFP è un obiettivo importante, soprattutto per alcune applicazioni, in cui la quantità della proteina o il tempo per la formazione del cromoforo sono fattori limitanti. Concettualmente, questo risultato può essere raggiunto per due strade diverse, ossia modificando le caratteristiche intrinseche di fluorescenza della proteina o agendo su altri parametri importanti nella produzione finale di un composto fluorescente. Per quanto riguarda le mutazioni del primo tipo, tra le quali la più fortunata è la sostituzione, all’interno del fluoroforo, della serina in posizione 65 con la treonina (mutazione S65T), esse cambiano il rapporto tra le due forme chimiche del cromoforo, e in particolare aumentano la concentrazione della forma ionizzata. Il risultato è che aumenta l’efficienza della GFP come fluoroforo eccitabile con luce blu, ossia nelle condizioni di illuminazione più adatte agli esperimenti con cellule vive, e nello stesso tempo quelle in cui la GFP risulta più stabile.
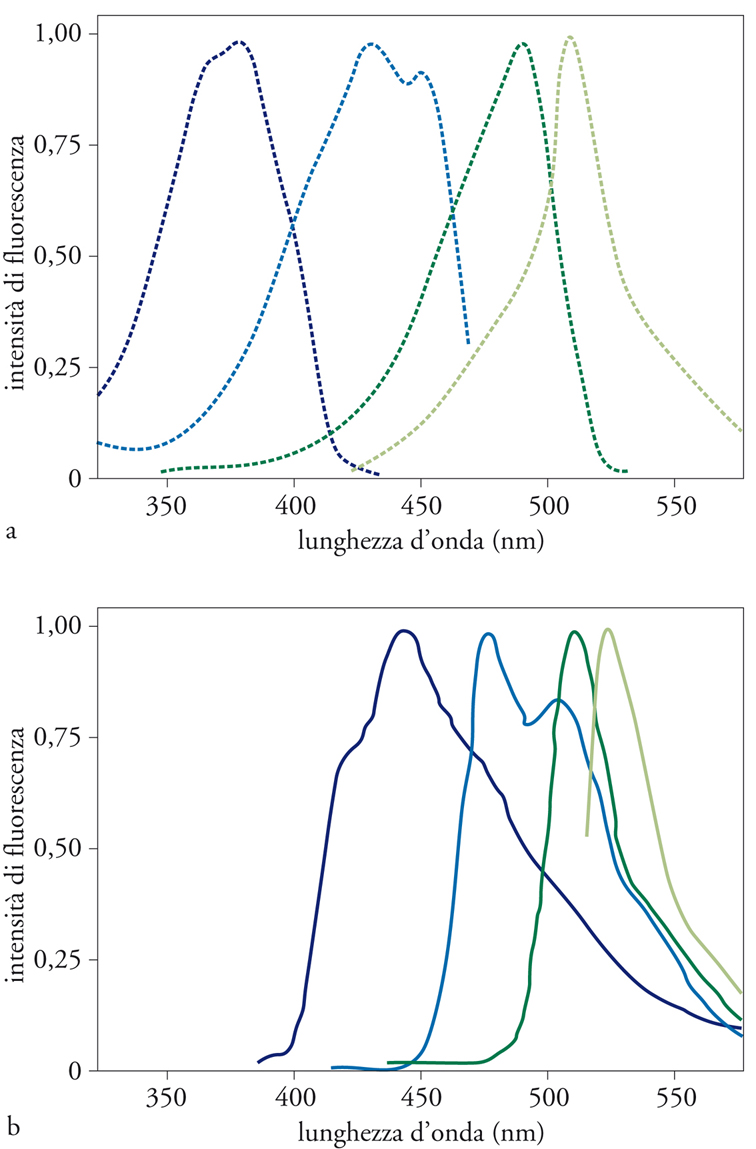
Nel caso del mutante S65T, il picco di eccitazione con luce UV è praticamente assente, mentre quello con luce blu è circa 6 volte maggiore che per la proteina nativa (fig. 6). Inoltre, la velocità di formazione del cromoforo di questo mutante è 4 volte maggiore di quella della forma nativa, e la forma attiva della proteina è 3,5 volte più resistente all’inattivazione da illuminazione (photobleaching). Per questi motivi, S65T è il mutante di elezione per esperimenti in cellula viva, e ha ormai largamente sostituito la forma nativa della GFP.
Le altre mutazioni, pur non modificando il cromoforo, e con esso le proprietà intrinseche di fluorescenza, interferiscono con tappe distinte del processo che porta alla formazione della proteina fluorescente. La prima serie di mutazioni, ormai di larga utilizzazione, è l’introduzione di mutazioni silenti che, non cambiando la sequenza della proteina, introducono nella sequenza del cDNA i codoni più comuni (ed efficienti) nelle cellule di mammifero, che spesso non coincidono con quelli di Aequorea. Questa procedura, che prende il nome di umanizzazione del cDNA, permette, a parità di mRNA (e quindi di efficienza di transfezione, di tempo intercorso ecc.), di produrre una quantità maggiore di proteina e raggiungere prima la soglia di visibilità al microscopio. Un secondo gruppo di mutazioni risolve due problemi associati alla produzione di questa proteina eterologa: il primo è la tendenza della proteina, quando sovraespressa, ad acquisire una conformazione errata e precipitare in corpi di inclusione non fluorescenti; il secondo consiste nella relativa inefficienza del processo di formazione del cromoforo a 37 °C (le condizioni sperimentali degli studi di biologia cellulare) rispetto a 15÷20 °C, la temperatura cui la proteina nativa è normalmente esposta. Infine, in alcuni tipi di piante (quali per es., Arabidopsis thaliana, un organismo modello ampiamente studiato), una mutazione silente che elimina un sito criptico di splicing presente nel cDNA della GFP aumenta marcatamente la quantità di proteina prodotta.
I mutanti con diversa lunghezza d’onda di eccitazione o di emissione. - Questi mutanti sono di grandissima utilità e ampliano considerevolmente le possibili applicazioni della GFP, in quanto possono essere impiegati, in associazione con la GFP nativa (o con il mutante S65T), per marcare simultaneamente più proteine o strutture di interesse, rivelare l’attività di due o più promotori oppure identificare, in animali transgenici, cellule di diversa derivazione embriologica.
Il requisito fondamentale perché un mutante di GFP possa essere utile a questo scopo è che la luce emessa possa essere facilmente distinta da quella nativa. Almeno tre mutanti spettrali soddisfano questi requisiti: il mutante blu, nel quale l’alterazione dello spettro è dovuta alla mutazione diretta del cromoforo (Y66H), che, eccitato con luce UV (380 nm), emette luce blu (445 nm); il mutante acquamarina, dovuto a una mutazione diversa dello stesso amminoacido (Y66W), e avente un picco di eccitazione bimodale nel violetto (433 e 453 nm) e di emissione tra il blu e il verde (con due picchi a 475 e 501 nm); il mutante giallo, dato dall’associazione della già descritta mutazione S65T con la T203Y, che, eccitato con luce verde (513 nm), emette luce gialla (picco a 527 nm). Oltre alle mutazioni responsabili dell’alterazione dello spettro, in questi mutanti sono introdotte altre mutazioni, che stabilizzano la struttura terziaria attorno al nuovo fluoroforo (nel caso, per esempio, del mutante blu, è presente la mutazione Y145F).
Le applicazioni della proteina GFP
La GFP come rivelatore dell’espressione genica. - Il controllo della distribuzione spaziale e temporale dell’espressione di un particolare gene risiede, di norma, nell’informazione contenuta nelle sequenze a monte della regione trascritta. La comprensione di questi meccanismi è un’area di ricerca molto intensa e un approccio frequentemente seguito è quello di frammentare la regione informativa e verificare la capacità delle diverse porzioni di indurre l’espressione di un gene reporter posto sotto il loro controllo trascrizionale. In altre parole, il cDNA della proteina reporter viene posto immediatamente a valle dell’ipotetico promotore da studiare; questo costrutto viene transfettato in cellule in coltura e la quantità di proteina espressa rivela l’efficacia del promotore. Le proteine normalmente utilizzate a questo scopo (doramfenicoloacetiltransferasi o CAT, β-galattosidasi o β-gal, luciferasi), poiché sfruttano la proprietà di convertire enzimaticamente substrati specifici in prodotti quantificabili, dipendono dall’interazione dell’enzima con il substrato e quindi, in cellule vive, dalla diffusibilità di quest’ultimo. Poiché i substrati attualmente disponibili diffondono con difficoltà attraverso le membrane biologiche, le cellule devono essere fissate o permeabilizzate per poter compiere l’indagine sperimentale. Con l’eccezione della luciferasi (che tuttavia, data la ridotta emissione di luce, richiede una strumentazione sofisticata e costosa), le proteine reporter finora disponibili non permettono di seguire dinamicamente il processo dell’espressione genica. La GFP, invece, grazie alla formazione autonoma del prodotto fluorescente e all’intensità del segnale, permette l’indagine a singola cellula e, in alcuni organismi modello trasparenti, come per esempio il nematode Caenorhabditis elegans, anche in animali transgenici. Tuttavia, a differenza delle proteine luminescenti (come la luciferasi), la GFP è contrastata dalla fluorescenza endogena delle cellule ed è quindi necessaria l’espressione di una quantità adeguata di prodotto fluorescente perché il segnale sia chiaramente riconoscibile. Per questo motivo, i mutanti brillanti descritti sopra sono indicati per questo tipo di applicazione, in cui si desidera rivelare precocemente l’attività di un promotore.
La GFP come marcatore di transfezione. - La modifica del patrimonio molecolare di una cellula con esperimenti di espressione di singole proteine tramite transfezione è un approccio efficace nello studio di processi cellula-ri complessi. Si pensi per esempio ai meccanismi che permettono alle cellule di riconoscere ligandi extracellulari (ormoni, fattori di crescita, trasmettitori) e tradurli in segnali intracellulari di attivazione di eventi, quali per esempio secrezione, motilità, differenziamento, crescita e così via. Essi sono costituiti nella maggior parte dei casi da vie complesse di segnalazione, spesso ridondanti, che convergono poi su effettori comuni. Per svelare le ragioni di questa complessità, e il ruolo specifico di proteine affini per struttura o funzione, si può esprimere una di queste molecole in forma nativa o come mutante con diverse proprietà biochimiche, e verificare l’effetto sulla funzione cellulare. Ciò può essere eseguito con due procedure: la selezione, dopo transfezione, di cloni cellulari che abbiano inserito nel genoma il gene esogeno e lo esprimano indefinitamente, o l’analisi in tempi brevi delle cellule transfettate, quando la frazione di cellule in cui è penetrato il plasmide esprime la proteina esogena. La prima procedura è più laboriosa e, analizzando la progenie di una sola cellula, rischia di enfatizzare differenze casuali, e non rappresentative, già contenute nella popolazione originaria. La seconda procedura analizza un campione di cellule più rappresentativo, ma per molti tipi di indagine è associata a un problema sperimentale importante: le cellule transfettate sono, a seconda del tipo cellulare e delle condizioni sperimentali, una frazione che varia dal 3 al 40%. Gli studi funzionali devono quindi essere eseguiti su questa percentuale di cellule, che deve essere riconosciuta dalle altre, non modificate rispetto alle cellule di partenza. Prima dell’avvento della GFP, si eseguiva l’analisi funzionaIe (per es., una misura di Ca2+ a singola cellula, o uno studio elettrofisiologico) ‘alla cieca’, poi si sacrificava la cellula e si verificava per immunocitochimica l’espressione della proteina eterologa. La GFP è in grado di risolvere questo problema, perché, se co-transfettata con il cDNA di interesse, svolge il ruolo di marcatore vitale delle cellule transfettate, che quasi invariabilmente saranno positive anche per l’altra proteina.
La GFP come marcatore di organuli e altre strutture cellulari. - L’organizzazione tridimensionale della cellula e dei suoi componenti è un importante argomento di studio, che le attuali tecniche di acquisizione e analisi di immagine permettono di affrontare con risoluzione elevata e qualità di immagine eccellente. La GFP è di grande ausilio in questo tipo di studi, perché, quando siano note le sequenze di indirizzamento specifiche, essa può essere selettivamente indirizzata a un organulo o compartimento di interesse tramite costrutti chimerici che comprendano il segnale di localizzazione opportuno. Avendo già trattato la costruzione delle chimere direzionate nel caso della fotoproteina equorina, non approfondiremo questo punto, salvo ricordare che la strategia e le sequenze impiegate sono le stesse usate per l’equorina.
Ci soffermeremo invece su un risultato sperimentale che fornisce un esempio chiaro dell’utilità di questo approccio. Le misure di Ca2+ mitocondriali ottenute con l’equorina ricombinante hanno dimostrato ampi aumenti della concentrazione di Ca2+ nella matrice, inaspettati sulla base delle caratteristiche biochimiche del trasportatore del Ca2+. L’ipotesi più plausibile per spiegare questa discrepanza è, come già discusso, che i mitocondri siano a stretto contatto con le sorgenti cellulari del Ca2+ (quale il reticolo endoplasmatico) e che quindi essi ‘sentano’ concentrazioni locali di Ca2+ molto elevate. Gli studi con GFP hanno permesso di dimostrare direttamente questa ipotesi. I mitocondri e il reticolo endoplasmatico sono stati marcati, in cellula viva, con due chimere di GFP distinte, ciascuna contenente un segnale di localizzazione, rispettivamente per i mitocondri e il reticolo endoplasmatico. I due organuli potevano essere riconosciuti tra loro perché le due chimere contenevano GFP con diverse caratteristiche spettrali: la GFP mitocondriale, o mtBFP, conteneva il mutante blu (Y66H), la GFP del reticolo endoplasmatico, o erGFP(S65T), il mutante verde brillante (S65T). Transfettando i due costrutti simultaneamente in cellule HeLa, è stato possibile ottenere separatamente l’immagine dei due organuli, illuminando prima il campione con luce UV (e raccogliendo l’emissione blu di mtBFP) e poi con luce blu (raccogliendo l’emissione verde di erGFP). Inoltre, allo scopo di ottenere un’immagine tridimensionale dei due organuli, indispensabile per apprezzare appieno le relazioni spaziali, sono state ottenute immagini progressive lungo l’asse verticale della cellula. Per minimizzare le distorsioni legate al movimento della cellula e degli organuli al suo interno è stato impiegato uno strumento sofisticato, che possiede un motore elettrico del piano del campione molto veloce, e una telecamera sensibilissima per raccogliere la luce emessa (e ridurre i tempi di esposizione a 10÷20 ms). In questo modo, è possibile completare la scansione verticale del campione in circa un secondo, un tempo in cui i movimenti cellulari sono molto modesti e non influenzano significativamente i dati ottenuti. Dopo l’acquisizione dell’immagine, speciali algoritmi permettono di rimuovere il segnale di fluorescenza che si origina dalle porzioni non a fuoco del campione, e quindi di ricostruire l’immagine tridimensionale. Il risultato finale è mostrato nella fig. 7. Esso si riferisce a una piccola porzione della cellula, e ha una risoluzione (80 nm) mai ottenuta in precedenza con altre sonde. Si può apprezzare il fatto che, come ci si attendeva, i due organuli formano reti complesse, le quali, in alcuni punti, vengono talmente a stretto contatto che le immagini non sono in grado di separarle.
La GFP come marcatore di specifiche proteine. - La distribuzione di una proteina all’interno di una cellula, e le sue variazioni dinamiche, sono spesso oggetto di indagine, per almeno due motivi. Il primo è che l’organizzazione spaziale di una cellula dipende strettamente dalla distribuzione dei suoi componenti molecolari, e quindi la comprensione dei meccanismi che sottendono all’indirizzamento intracellulare delle proteine è un obiettivo importante. Si pensi, per esempio, al traffico delle proteine attraverso la membrana nucleare o alla progressione delle proteine lungo la via secretoria, due argomenti di grande attualità. In questi studi, poter seguire direttamente una proteina nativa, o un mutante in cui le sequenze che si ritengono importanti per l’indirizzamento siano state mutate, è un approccio molto efficace. Ma vi è anche almeno un altro campo di indagine che può beneficiare dalla possibilità di seguire dinamicamente la localizzazione di una proteina: lo studio dei meccanismi con cui i segnali intracellulari vengono tradotti all’interno della cellula. È oggi noto, infatti, che, con meccanismi differenti, stimoli esterni assai diversi tra loro, quali ormoni steroidei e agonisti che inducono la scissione del fosfatidilinositolo bisfosfato (PIP2) in diacilglicerolo (DAG) e inositolotrisfosfato (IP3), inducono la traslocazione intracellulare di proteine segnale.
In particolare, gli ormoni steroidei inducono la traslocazione al nucleo del loro recettore che può quindi agire sul promotore dei geni responsivi, mentre gli aumenti dei livelli di concentrazione di Ca2+ e di DAG indotti da agonisti inducono la traslocazione in membrana delle diverse isoforme di proteinchinasi C (PKC, Protein kinase C). Poter seguire il destino intracellulare di queste proteine significa quindi seguire dinamicamente lo stato di attivazione di una cellula e i meccanismi con cui viene indotto.
L’esperimento illustrato in fig. 8 è un esempio tipico di tale applicazione. In questo studio è stata utilizzata una chimera di GFP fusa con la isoforma δ della proteinchinasi C. La transfezione di questa chimera dota la cellula di una PKCδ direttamente visibile all’indagine microscopica. La chimera PKCδ/GFP in una cellula a riposo mostra una chiara localizzazione citoplasmatica (e infatti in un altro esperimento, non mostrato, in cui la membrana plasmatica viene permeabilizzata con digitonina, la chimera viene completamente rilasciata). In seguito a stimolazione con esteri del forbolo – agenti cancerogeni che mimano il comportamento del DAG, e quindi inducono il loro effetto biologico agendo sulla via di segnalazione DAG/Ca2+ – la PKC viene quasi interamente traslocata alla membrana plasmatica, dove può agire su effettori specifici e causare la risposta biologica. La chimera PKC/GFP permette di seguire in tempo reale, e in cellula viva, questo importante processo biologico.
Bibliografia
Allen 1977: Allen, David G. - Blinks, John R. - Prendergast, Franklin G., Aequorin luminescence: relation of light emission to calcium concentration - a calcium-independent component, “Science”, 195, 1977, pp. 996-998.
Chalfie 1994: Chalfie, Martin e altri, Green fluorescent protein as a marker for gene expression, “Science”, 263, 1994, pp. 802-805.
Dobbie 2008: Dobbie, Ian M. - Lowndes, Noel F. - Sullivan, Kevin F., Autofluorescent proteins, “Methods in cell biology”, 85, 2008, pp. 1-22.
Dupriez 2002: Dupriez, Vincent J. e altri, Aequorin-based functional assays for G-protein-coupled receptors, ion channels, and tyrosine kinase receptors, “Receptors and channels”, 8, 2002, pp. 319-330.
Haseloff, Siemering 2006: Haseloff, Jim - Siemering, Kirby R., The uses of green fluorescent protein in plants, “Methods of biochemical analysis”, 47, 2006, pp. 259-284.
Heim 1995: Heim, Roger - Cubitt, Andrew B. - Tsien, Roger Y., Improved green fluorescence, “Nature”, 373, 1995, pp. 663-664.
Inouye 1985: Inouye, Satoshi e altri, Cloning and sequence analysis of cDNA for the luminescent protein aequorin, “Proceedings of the National Academy of Sciences USA”, 82, 1985, pp. 3154-3158.
Ormo 1996: Ormo, Mats e altri, Crystal structure of the Aequorea victoria green fluorescent protein, “Science”, 273, 1996, pp. 1392-1395.
Pinton 2007: Pinton, Paolo e altri, Biosensors for the detection of calcium and pH, “Methods in cell biology”, 80, 2007, pp. 297-325.
Pozzan 2003: Pozzan Tullio - Mongillo, Marco - Rudolf, Rüdiger, The Theodore Bücher lecture. Investigating signal transduction with genetically encoded fluorescent probes, “European journal of biochemistry”, 270, 2003, pp. 2343-2352.
Querido, Chartrand 2008: Querido, Emmanuelle - Chartrand, Pascal, Using fluorescent proteins to study mRNA trafficking in living cells, “Methods in cell biology”, 85, 2008, pp. 273-292.
Rudolf 2003: Rudolf, Rüdiger e altri, Looking forward to seeing calcium, “Nature reviews. Molecular cell biology”, 4, 2003, pp. 579-586.
Shaner 2007: Shaner, Nathan C. - Patterson, George H. - Davidson, Michael W., Advances in fluorescent protein technology, “Journal of cell science”, 120, 2007, pp. 4247-4260.
Shimomura 2005: Shimomura, Osamu, The discovery of aequorin and green fluorescent protein, “Journal of microscopy”, 217, 2005, pp. 1-15.
Straight 2007: Straight, Aaron F., Fluorescent protein applications in microscopy, “Methods in cell biology”, 81, 2007, pp. 93-113.
Tsien 1982: Tsien, Roger Y. - Pozzan, Tullio - Rink, Timothy J., T cell mitogens cause early changes in cytoplasmic free Ca2+ and membrane potential in lymphocytes, “Nature”, 295, 1982, pp. 68-71.
Ward 2006: Ward, William W., Biochemical and physical properties of green fluorescent protein, “Methods of biochemical analysis”, 47, 2006, pp. 39-65.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


